CH4/H2O/CO2在β-SiO2(100)面吸附的第一性原理研究
2019-09-17 11:36:06趙建飛汪周華
原子與分子物理學(xué)報(bào) 2019年4期
關(guān)鍵詞:研究
趙建飛, 汪周華, 郭 平, 羅 強(qiáng)
(1. 西南石油大學(xué)油氣藏地質(zhì)及開(kāi)發(fā)工程國(guó)家重點(diǎn)實(shí)驗(yàn)室,成都 610500; 2. 西南石油大學(xué)理學(xué)院,成都 610500)
1 引 言
作為全球三大非常規(guī)天然氣之一,頁(yè)巖氣在全世界范圍內(nèi)受到了廣泛的重視,美國(guó)和加拿大已經(jīng)實(shí)現(xiàn)了頁(yè)巖氣的商業(yè)開(kāi)采[1],并取得較好成果,我國(guó)在頁(yè)巖氣的研究現(xiàn)狀、成藏機(jī)理和有利區(qū)評(píng)價(jià)方面做了大量工作,但近年才開(kāi)始研究頁(yè)巖氣的吸附現(xiàn)象[2, 3]. 頁(yè)巖氣主要由CH4、C2H6、CO2、N2等氣體組成,其中CH4含量可達(dá)95%以上[4],其賦存介質(zhì)為頁(yè)巖,通常在潮汐沼澤或者深水盆地的水環(huán)境下沉積,水充填在有機(jī)質(zhì)孔隙及無(wú)機(jī)礦物孔隙中[5]. 頁(yè)巖的礦物組成以石英為主,部分地區(qū)石英含量在35.7%~92.6%之間[6]. 根據(jù)不同氣體的吸附能力不同,許多學(xué)者提出頁(yè)巖氣藏注二氧化碳提高采收率[7],因此,研究CH4、CO2和H2O在頁(yè)巖主要礦物成分SiO2表面的吸附,對(duì)了解頁(yè)巖氣的真實(shí)賦存狀態(tài)、定量評(píng)估氣體的吸附能力、注氣開(kāi)采等方面具有重要意義.
研究者們采用大量的實(shí)驗(yàn)和理論研究了頁(yè)巖氣與頁(yè)巖基質(zhì)的吸附作用[8-10],研究表明頁(yè)巖氣主要吸附于干酪根和黏土礦物中,很少研究石英表面的吸附. 吉利明等[4]采用等溫吸附實(shí)驗(yàn)方法,對(duì)不同來(lái)源和成因的泥頁(yè)巖中的常見(jiàn)黏土礦物進(jìn)行了甲烷吸附實(shí)驗(yàn),結(jié)果表明不同類(lèi)型的黏土礦物的氣體吸附能力有明顯差異,但無(wú)直接證據(jù)表明氣體在石英表面的吸附性,而部分學(xué)者研究認(rèn)為,吸附氣量與石英含量呈正相關(guān)性[11],故石英表面吸附有待深入研究. 常規(guī)油氣向頁(yè)巖的發(fā)展其實(shí)就是微米空間向納米空間的發(fā)展,宏觀(guān)上突破越來(lái)依賴(lài)于微觀(guān)成果,因而采用新理論方法加強(qiáng)微觀(guān)研究顯得尤為重要[12]. 應(yīng)用量子力學(xué)理論的第一性原理計(jì)算是從電子結(jié)構(gòu)出發(fā),只借助于基本常量和合理近似來(lái)進(jìn)行的計(jì)算方法,它能夠從電子和原子層面認(rèn)識(shí)材料的物理性質(zhì),近年來(lái),利用第一性原理在CH4的吸附研究中取得很多有價(jià)值的研究成果[13]. 李萍等[14]采用基于密度泛函理論的廣義梯度近似方法(GGA/PE91)研究了甲烷在Ni(110)表面不同高對(duì)稱(chēng)位的吸附行為,研究結(jié)果表明,CH4在Ni(110)表面頂位T4的吸附最穩(wěn)定,吸附能為4.59 kJ/mol,為物理吸附. Rodríguez-Kessler等[15]用GGA-PBE泛函研究了CH4在最穩(wěn)定Nin(2-16,21,55)團(tuán)簇表面的吸附,研究結(jié)果表明CH4在Ni4團(tuán)簇結(jié)構(gòu)的頂位吸附時(shí)最穩(wěn)定,吸附能為-0.47 eV. Zhu等[16]采用DFT-D3的方法研究了甲烷在頁(yè)巖中干酪根表面的吸附機(jī)制,結(jié)果表明,甲烷與干酪根之間的相互作用主要是范德瓦爾斯力,其吸附能只有14 kJ/mol左右. 王三躍等[17]采用GGA/PW91泛函研究了甲烷在MOF-5金屬有機(jī)骨架中不同吸附位吸附的結(jié)構(gòu)模型,并計(jì)算了甲烷的吸附能,甲烷的吸附與含氧極性官能團(tuán)的朝向有關(guān),當(dāng)碳?xì)滏I指向氧位時(shí),甲烷具有最優(yōu)吸附結(jié)構(gòu),同時(shí),在骨架中引入電子基團(tuán)可以增強(qiáng)MOFs對(duì)甲烷的吸附作用. Luo等人[18]基于密度泛函理論的第一性原理方法,研究了頁(yè)巖氣在CaCO3(100)面吸附的結(jié)構(gòu)和電子性質(zhì),其對(duì)致密氣在石英砂巖表面吸附性強(qiáng)弱的論證[19]為本研究提供了借鑒. 通過(guò)前人的研究發(fā)現(xiàn),在第一性原理計(jì)算研究中,從頁(yè)巖氣藏注二氧化碳提高采收率角度以及含水角度出發(fā)研究頁(yè)巖氣在其主要無(wú)機(jī)礦物上的吸附較少,基于此,采用密度泛函理論研究CH4、H2O、CO2在β-SiO2表面不同位置吸附的穩(wěn)定性,對(duì)比分析其吸附狀況,進(jìn)而研究其吸附作用的吸附能、態(tài)密度等微觀(guān)機(jī)理.
2 計(jì)算方法及過(guò)程
2.1 計(jì)算方法
使用基于密度泛函理論(Density Function Theory,DFT)[20, 21]的第一性原理計(jì)算方法研究CH4、H2O和CO2氣體在β-SiO2表面不同位置吸附的幾何結(jié)構(gòu)和電子特性,對(duì)比分析其在不同高對(duì)稱(chēng)位的吸附狀況. 計(jì)算使用的CASTEP[22, 23]是一種從量子力學(xué)角度對(duì)固體材料進(jìn)行理論模擬研究的從頭算量子力學(xué)程序,由英國(guó)劍橋大學(xué)凝聚態(tài)理論研究組開(kāi)發(fā). 模擬計(jì)算時(shí),采用Perdew等[24]提出的廣義梯度近似(Generalized Gradient Approximation,GGA)中的PBE泛函處理交換關(guān)聯(lián)勢(shì),同時(shí)各收斂精度設(shè)為:?jiǎn)卧幽芰渴諗烤?.0×10-5eV/atom,作用于單原子上的最大力收斂精度0.05 eV/?,最大壓力收斂精度0.1 GPa,最大位移收斂精度0.002 ?. 同時(shí),采用Broyden等提出的BFGS優(yōu)化算法[25-28]進(jìn)行優(yōu)化,平面波截止能測(cè)試后選取380 eV,SCF自洽精度設(shè)為單原子能量收斂至2.0×10-6eV,K點(diǎn)[29]網(wǎng)格尺寸采用1×1×1.
2.2 理論模型
采用Materials Studio 2016數(shù)據(jù)庫(kù)提供的SiO2_quartz_beta模型,晶格常數(shù)為a=b=5.01 ?,c=5.47 ?,α=β=90°,γ=120°,優(yōu)化后建立p(2×2)超晶胞作為初始模型并截取(100)面,建立厚度20 ?的真空層,模擬β-SiO2(100)面并對(duì)其繼續(xù)結(jié)構(gòu)優(yōu)化. 為減少運(yùn)算量以及弱化基底厚度影響,吸附計(jì)算前弛豫表面,即對(duì)表面Si原子以外的原子進(jìn)行固定而表層硅原子與吸附質(zhì)在結(jié)構(gòu)優(yōu)化計(jì)算中位置可變. 吸附質(zhì)與表面的距離試算后設(shè)為4 ?,在覆蓋度θS[30]為0.25ML(monolayer)的條件下對(duì)吸附情況進(jìn)行研究.
β-SiO2(100)面表面層由四個(gè)Si原子構(gòu)成一個(gè)矩形,[100]方向邊長(zhǎng)為5.087 ?,[010]方向邊長(zhǎng)為5.580 ?. 吸附質(zhì)分子在β-SiO2(100)面的高對(duì)稱(chēng)吸附位有頂位(T位)、長(zhǎng)橋位(LB位)、短橋位(SB位)、表面四重洞位(H位),圖1為高對(duì)稱(chēng)吸附位示意圖. 為利用吸附質(zhì)分子結(jié)構(gòu)的對(duì)稱(chēng)性,CH4以碳原子為中心,兩個(gè)氫原子在下等高,另外兩個(gè)氫原子在上等高;H2O以氧原子為中心,兩個(gè)氫原子在下等高;CO2以碳原子為中心三個(gè)原子等高且平行于吸附面.
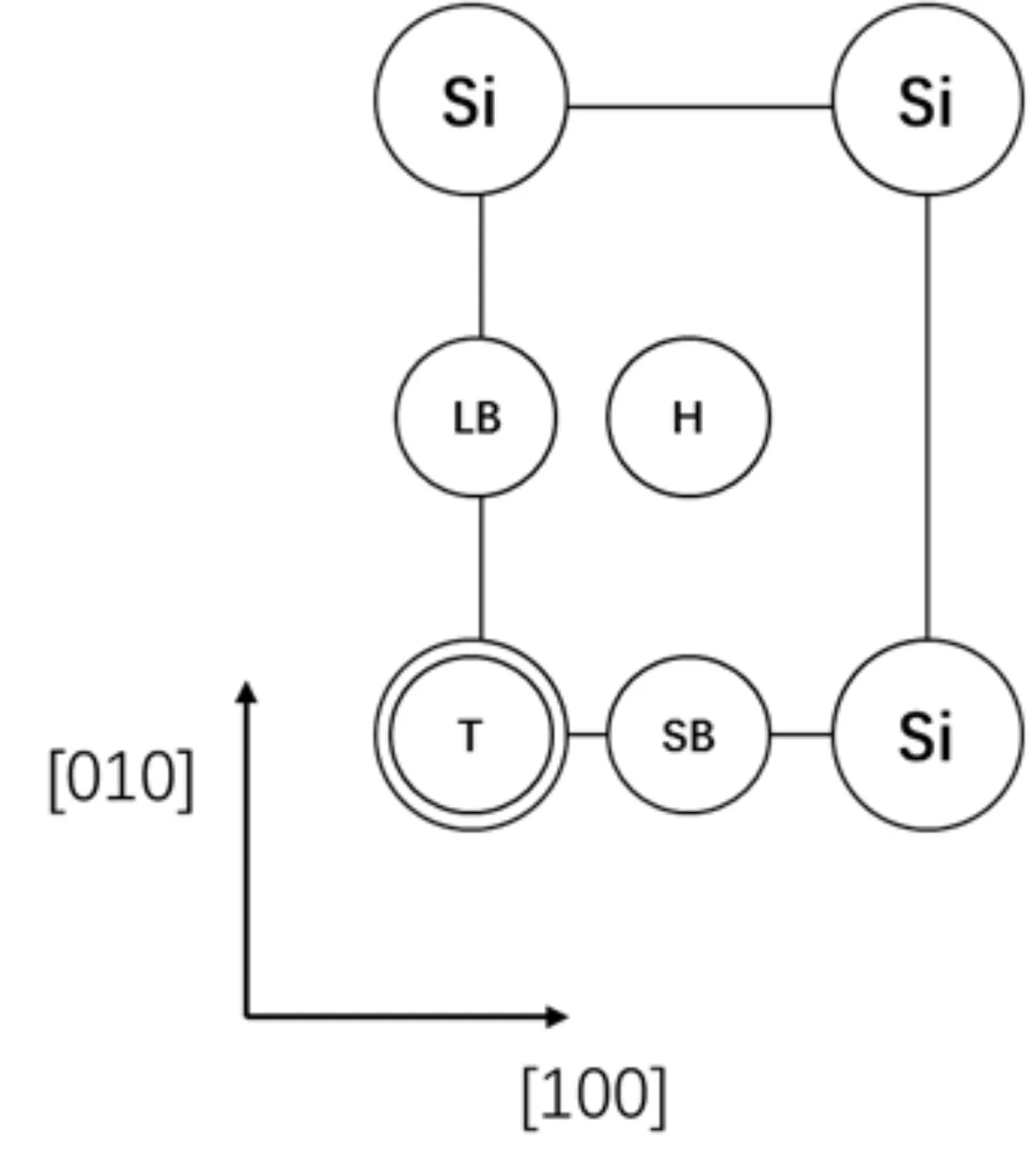
圖1 高對(duì)稱(chēng)吸附位:T、H、SB與LBFig.1 Adsorption sites: T, H, SB and LB
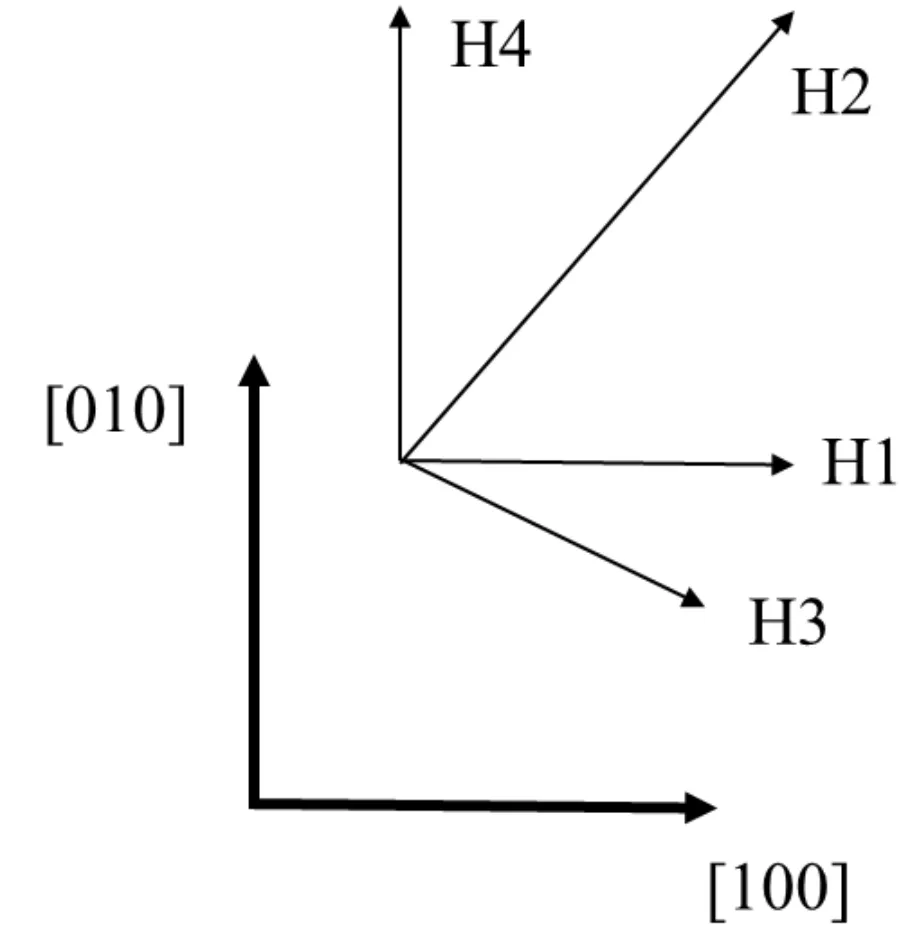
圖2 H位四個(gè)吸附方向Fig.2 Four adsorption directions of H site
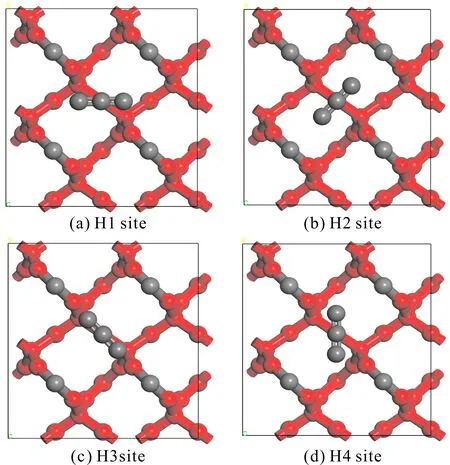
圖3 H位吸附方式示意圖(紅色:被固定的原子,灰色:未被固定的原子)Fig. 3 Adsorption modes of H sites (red: fixed atoms, gray: unfixed atoms)
3 計(jì)算結(jié)果和分析
3.1 吸附質(zhì)在β-SiO2(100)面上吸附統(tǒng)計(jì)
吸附能表示吸附前后體系總能量的變化,計(jì)算公式如下:
Ead=EAdsorbate/SiO2-(EAdsorbate+ESiO2)
(1)
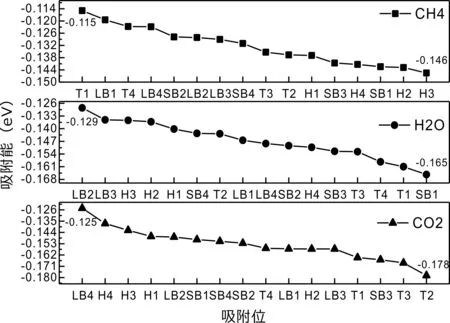
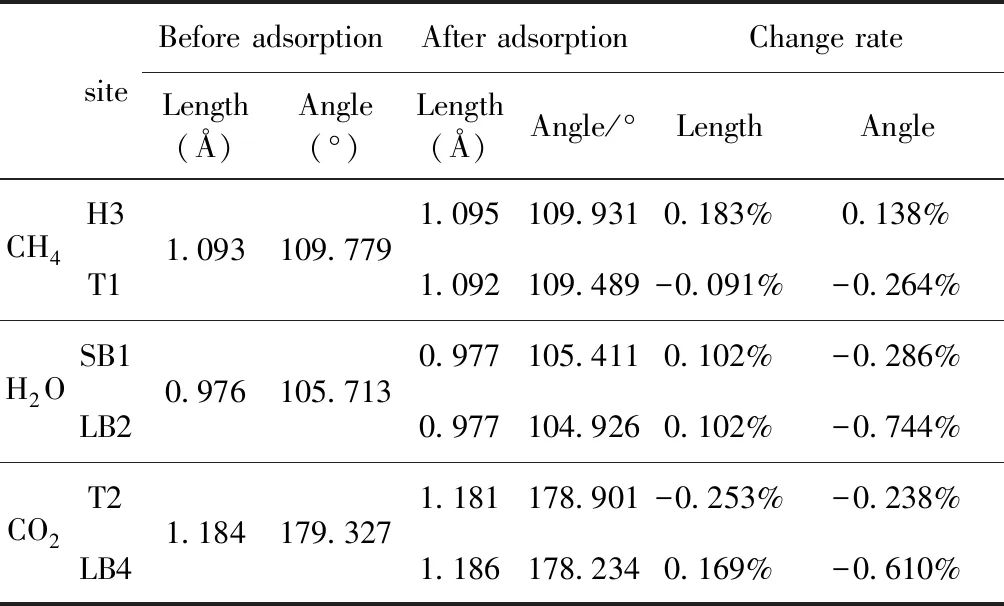
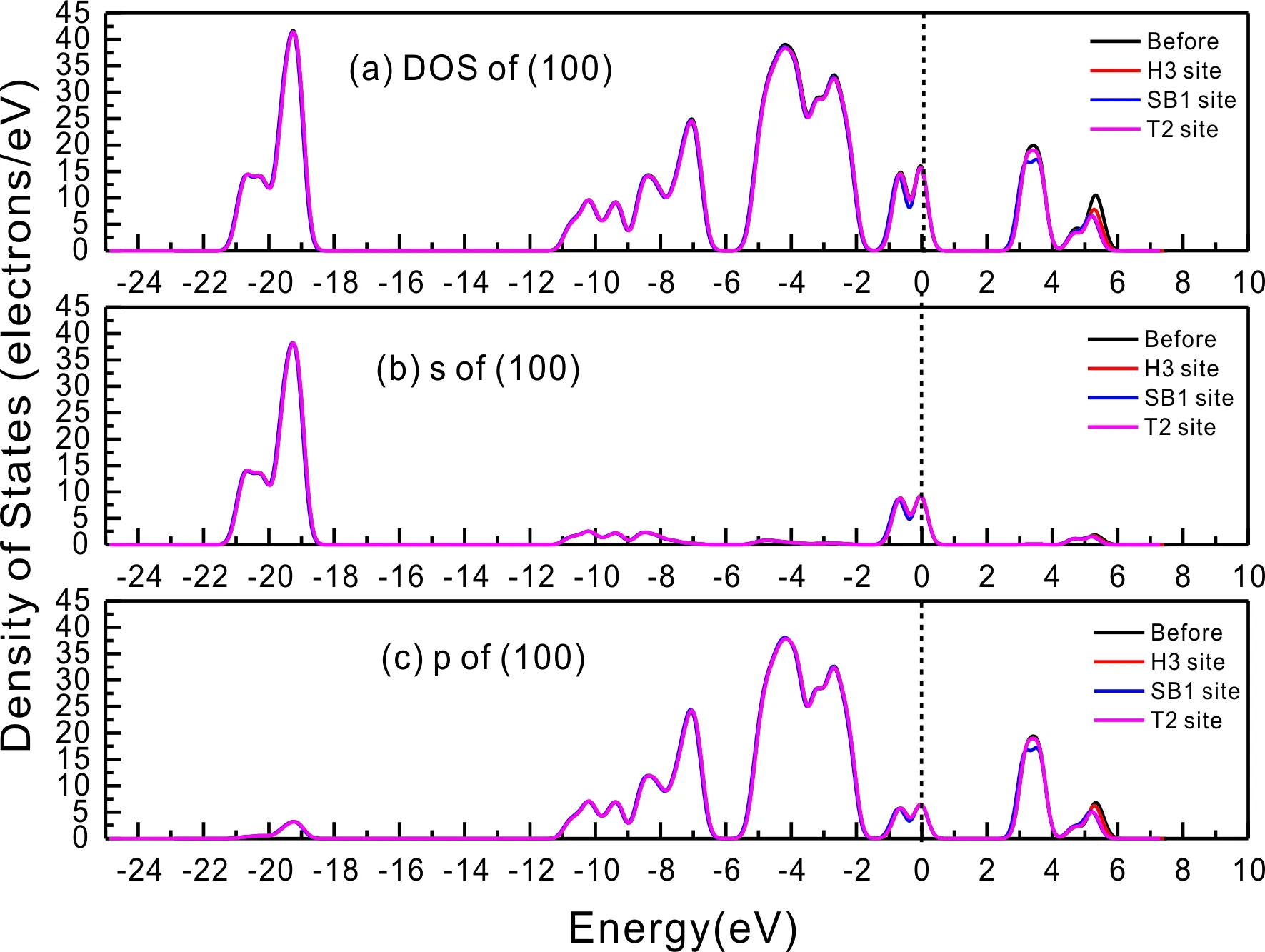
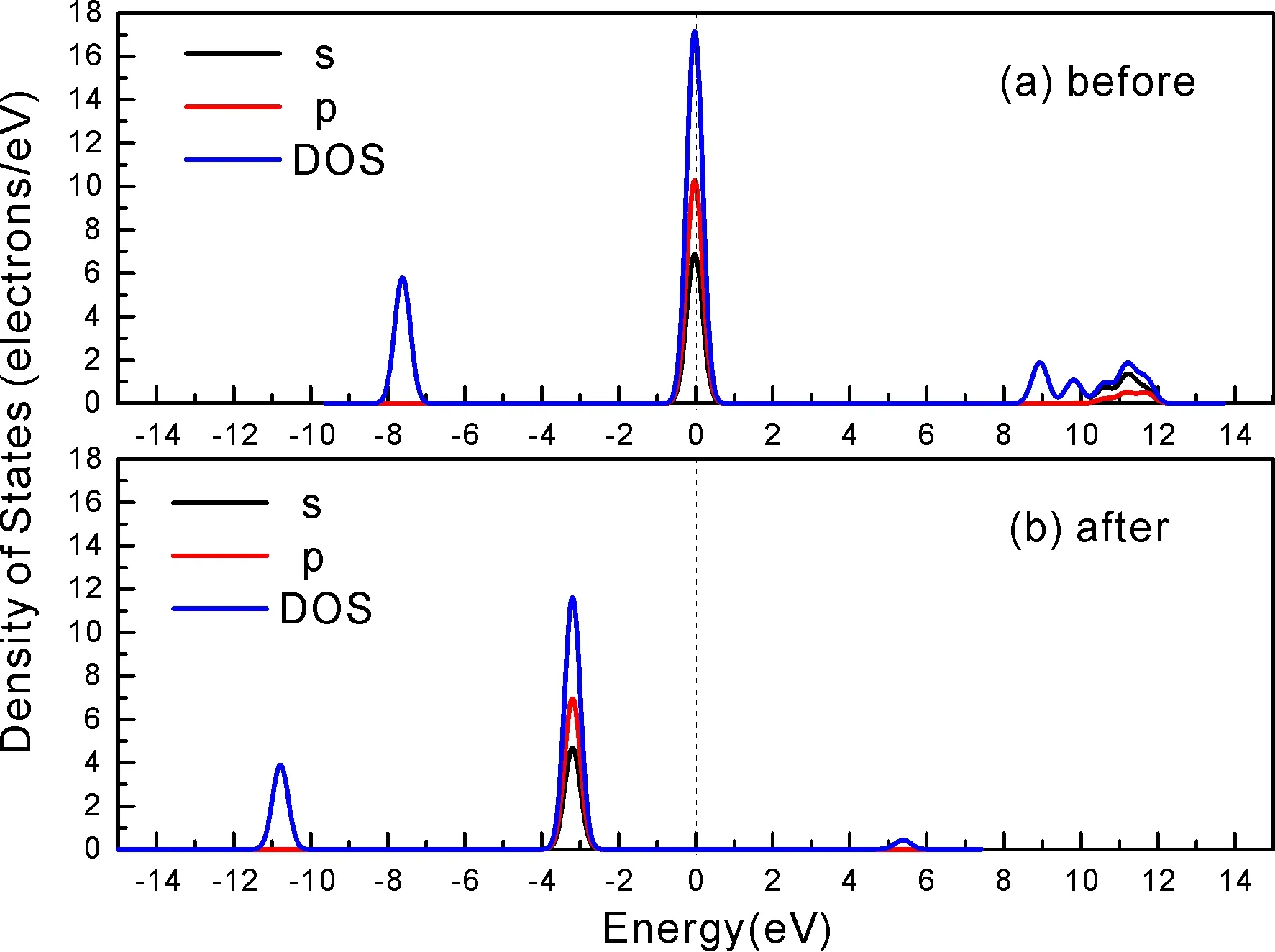
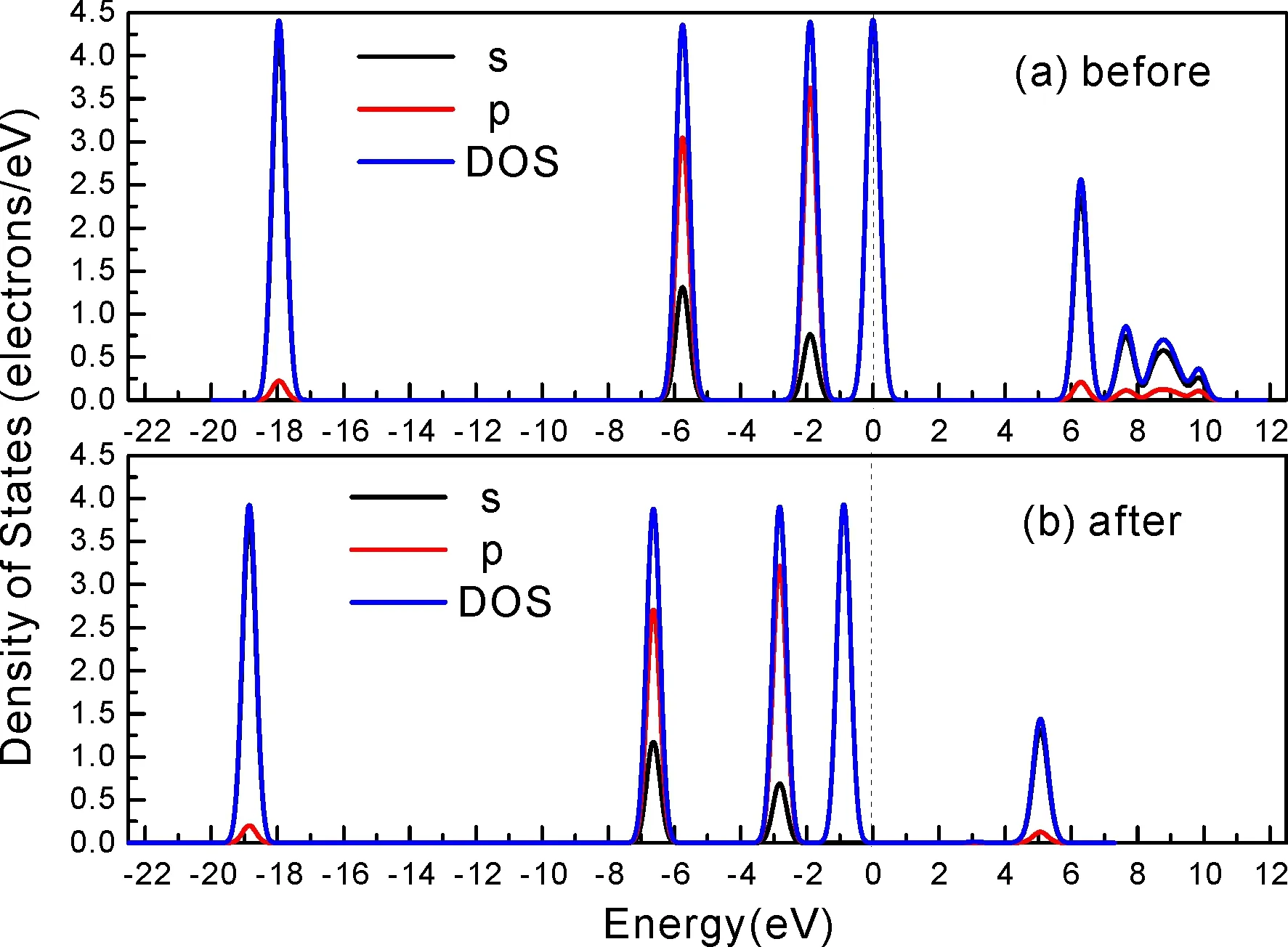
式中,Ead表示各吸附位的吸附能;EAdsorbate/SiO2表示吸附質(zhì)吸附在β-SiO2(100)面時(shí)體系的總能量;EAdsorbate表示各吸附質(zhì)的能量;ESiO2表示β-SiO2(100)面吸附前的能量. 吸附能計(jì)算值為負(fù)時(shí)表明發(fā)生放熱反應(yīng)且吸附后體系更加穩(wěn)定,Ead越小表示體系的穩(wěn)定性越強(qiáng);反之,Ead越大表示體系的穩(wěn)定性越差. 吸附能滿(mǎn)足-0.62 eV 表1 CH4、H2O和CO2在β-SiO2表面的吸附能(eV) Table 1 Adsorption energies (eV) of adsorbate onβ-SiO2(100) surface TEad,HEad,LBEad,SBEad,ⅠT1-0.115 H1-0.137 LB1-0.119 SB1-0.142 T2-0.137 H2-0.143 LB2-0.128 SB2-0.128 T3-0.135 H3-0.146 LB3-0.129 SB3-0.141 T4-0.123 H4-0.141 LB4-0.123 SB4-0.131 ⅡT1-0.161 H1-0.140 LB1-0.146 SB1-0.165 T2-0.143 H2-0.136 LB2-0.129 SB2-0.149 T3-0.153 H3-0.135 LB3-0.135 SB3-0.153 T4-0.158 H4-0.150 LB4-0.148 SB4-0.143 ⅢT1-0.164 H1-0.147 LB1-0.157 SB1-0.150 T2-0.178 H2-0.157 LB2-0.148 SB2-0.153 T3-0.168 H3-0.142 LB3-0.157 SB3-0.166 T4-0.157 H4-0.137 LB4-0.125 SB4-0.151 由表1可知,CH4、H2O和CO2在β-SiO2(100)面不同吸附位的吸附能分布在-0.2 eV ~ -0.1 eV區(qū)間內(nèi)(1 eV=96.4853 kJ/mole[32]),均大于-0.62 eV且小于0 eV,為物理吸附. 根據(jù)上表數(shù)據(jù)做出吸附能隨吸附位由大到小排列的關(guān)系曲線(xiàn)圖,如圖4所示. 圖4 吸附質(zhì)在β-SiO2(100)面吸附能隨吸附位變化曲線(xiàn)Fig. 4 Adsorption energy curves of adsorbates on the surface of β-SiO2 由圖4可直觀(guān)的看出,CH4在β-SiO2(100)面H3位吸附能最小,為-0.146 eV,表示CH4在該位吸附最穩(wěn)定,而在T1位吸附能最大,為-0.115 eV,即CH4在T1位吸附最不穩(wěn)定,吸附能遠(yuǎn)低于40 kJ/mole,表明CH4在β-SiO2(100)表面的吸附為物理吸附,與文獻(xiàn)[33]研究結(jié)論一致. H2O在SB1位吸附能最小且為-0.165 eV,而在LB2位吸附能最大且為-0.129 eV. CO2在T2位吸附能最小且為-0.178 eV,而在LB4位吸附能最大,為-0.125 eV. 不同吸附質(zhì)最小吸附能大小依次為: CH4> H2O> CO2,即,CO2的吸附能力最強(qiáng),H2O次之,CH4最弱. 同時(shí),各吸附質(zhì)不同吸附位的吸附能變化范圍非常狹窄,最大吸附能與最小吸附能分別相差0.031 eV、0.036 eV、0.053 eV,表明不同吸附位對(duì)吸附質(zhì)分子的吸附影響較小,導(dǎo)致其在表面上容易發(fā)生穩(wěn)定流動(dòng)[19],但相對(duì)而言β-SiO2(100)面優(yōu)先吸附CO2. 對(duì)比分析吸附質(zhì)吸附前后以及吸附能最大與最小時(shí)的幾何結(jié)構(gòu)變化有利于理解吸附作用強(qiáng)弱,表2列出了吸附質(zhì)在最穩(wěn)定吸附位與最不穩(wěn)定位吸附前后的物理結(jié)構(gòu),變化率為正表示鍵長(zhǎng)伸長(zhǎng)或鍵角增大,為負(fù)表示鍵長(zhǎng)縮短或鍵角減小. 由表2可知,CH4在H3位吸附后鍵長(zhǎng)變化率為0.183%,鍵角變化率為0.138%;而CH4在T1位吸附后鍵長(zhǎng)變化率為-0.091%,鍵角變化率為-0.264%;H2O在SB1位吸附后鍵長(zhǎng)變化率為0.102%,鍵角變化率為-0.286%;而在LB2位吸附后鍵長(zhǎng)變化率為0.102%,鍵角變化率為-0.744%;CO2在T2位吸附后鍵長(zhǎng)變化率為-0.253%,鍵角變化率為-0.238%;而在LB4位吸附后鍵長(zhǎng)變化率為0.169%,鍵角變化率為-0.610%. 吸附能最大時(shí)吸附質(zhì)的鍵角絕對(duì)變化率均大于吸附能最小時(shí)的,吸附質(zhì)的物理結(jié)構(gòu)變化微弱表明其所受作用力微弱[14],[34]. 表2 吸附質(zhì)吸附前后的物理結(jié)構(gòu) Table 2 Physical structures of adsorbate before and after adsorption siteBefore adsorptionAfter adsorptionChange rateLength(?)Angle(°)Length(?)Angle/°LengthAngleCH4H3T11.093109.7791.095109.9310.183%0.138%1.092109.489-0.091%-0.264%H2OSB1LB20.976105.7130.977105.4110.102%-0.286%0.977104.9260.102%-0.744%CO2T2LB41.184179.3271.181178.901-0.253%-0.238%1.186178.2340.169%-0.610% 借助態(tài)密度(Density of States,DOS)分析可以進(jìn)一步理解吸附質(zhì)在β-SiO2(100)面的吸附作用,分態(tài)密度(Partial Density of States,PDOS)可以分析體系吸附后原子的軌道對(duì)態(tài)密度的貢獻(xiàn),s分態(tài)密度由體系各原子不同s軌道雜化構(gòu)成,p分態(tài)密度由p軌道雜化構(gòu)成. 圖5所示為各吸附質(zhì)最穩(wěn)定吸附位體系的態(tài)密度圖,由圖可知,態(tài)密度曲線(xiàn)在-23.2~ -21.2、-11.1~ -10.3、-7.0~ -6.0、-3.6~ -2.7,-1.1 eV~ -0.4、2.7~ 6.0 eV區(qū)間內(nèi)存在差異,其余能量區(qū)間態(tài)密度重合. 其中,能量在-23.2 eV~ -21.2 eV區(qū)間內(nèi)態(tài)密度為T(mén)2位獨(dú)有,由s分態(tài)密度貢獻(xiàn);-7.0 eV~ -6.0 eV區(qū)間內(nèi)T2位態(tài)密度均大于H3、SB1位,由p分態(tài)密度貢獻(xiàn);-3.6 eV~ -2.7 eV區(qū)間內(nèi)H3位態(tài)密度左移,由sp分態(tài)密度共同作用,T2位態(tài)密度大于SB1位態(tài)密度則由p分態(tài)密度貢獻(xiàn). 各吸附質(zhì)最穩(wěn)定吸附位態(tài)密度高度重合表明各吸附質(zhì)與β-SiO2表面相互作用相似且差異較小. 圖5 H3、SB1 與T2位態(tài)密度Fig. 5 DOSs of H3、SB1 and T2 sites 圖6 (100)面吸附前后態(tài)密度Fig. 6 DOSs of (100) surface before and after adsorption 圖6為吸附質(zhì)最穩(wěn)定吸附位吸附前后基底面的態(tài)密度,從圖中可以看出,吸附前與吸附后表面的態(tài)密度曲線(xiàn)基本重合,僅在能量為3.3~ 3.6、5.1~ 5.7 eV區(qū)間有較小差異,表明不同吸附質(zhì)吸附對(duì)β-SiO2(100)表面DOS影響很小. 對(duì)比s和p分態(tài)密度可以看出,表面的DOS由s、p共同貢獻(xiàn),但各自有明顯的界限,低能量區(qū)(-21.3 eV~ -18.6 eV)時(shí),s分態(tài)密度對(duì)DOS起主要作用,高能量區(qū)則p對(duì)DOS起主要作用,吸附前后PDOS分別與吸附前基本重合,進(jìn)一步說(shuō)明吸附作用對(duì)表面影響小,吸附質(zhì)在β-SiO2(100)面的吸附非常微弱,為進(jìn)一步理解吸附作用對(duì)吸附質(zhì)影響,對(duì)CH4、H2O和CO2各自進(jìn)行吸附前后態(tài)密度對(duì)比分析. 圖7 CH4吸附前后態(tài)密度Fig. 7 DOSs of CH4 before and after adsorption 圖7為H3位吸附前后CH4態(tài)密度,由圖可知,CH4的態(tài)密度由s態(tài)和p態(tài)電子貢獻(xiàn). 對(duì)比吸附前后態(tài)密度分布可知,吸附前態(tài)密度分布在-8.1~ -7.0、-0.7~ 0.5與8.4~ 12.1 eV區(qū)間,吸附后主要分布在-11.2~ -10.3和-3.8~ -2.8 eV,吸附后CH4的態(tài)密度整體向更低能量區(qū)域偏移大約3.2 eV且態(tài)密度峰值降低,吸附后能量降低結(jié)構(gòu)更加穩(wěn)定,原8.4 eV~12.1 eV處的態(tài)密度幾乎消失.s分態(tài)密度由吸附前0 eV處的6.875 electrons/ eV,-7.6 eV處的5.785 electrons/eV變?yōu)槲胶?3.2 eV處的4.662 electrons/eV,-10.8 eV的3.886 electrons/eV,降幅約2.2 electrons/eV,p分態(tài)密度由吸附前0 eV的10.269 electrons/eV變?yōu)槲胶?3.2 eV的6.943 electrons/eV,降幅約3.3 electrons/eV. 吸附后電子態(tài)密度峰值的數(shù)量減少,且峰值均有不同程度的下降,說(shuō)明吸附作用對(duì)CH4的電子態(tài)密度的分布有著較顯著的影響. 圖8 H2O吸附前后態(tài)密度Fig. 8 DOSs of H2O before and after adsorption SB1位吸附H2O前后的態(tài)密度如圖8所示,吸附后H2O的態(tài)密度整體向低能量區(qū)域偏移約0.8 eV,且態(tài)密度峰值由吸附前多個(gè)主要峰值降為5個(gè)主要峰值,除高能量區(qū)變化較為明顯外,低能區(qū)峰值變化較小,說(shuō)明吸附后電子更多的占據(jù)低能量的能態(tài),吸附后較吸附前的結(jié)構(gòu)更穩(wěn)定. 圖9 CO2吸附前后態(tài)密度Fig. 9 DOSs of CO2 before and after adsorption 圖9所示為T(mén)2位吸附CO2前后的態(tài)密度,吸附前態(tài)密度分布在-20.6~ -18.5、-5.3~ -3.1、-0.6~ 0.4與7.6~10.8 eV區(qū)間內(nèi),吸附后主要分布在-23.5~ -21.4、-8.0~ -5.9和-3.7~ -2.1 eV,吸附后態(tài)密度曲線(xiàn)整體向低能區(qū)移動(dòng)3.0 eV,峰值具有不同幅度降低,從低能量區(qū)到高能量區(qū)各峰值分別由4.392、4.402、4.401、12.839、8.856、9.266 electrons/eV變?yōu)?.916、3.904、3.913、11.579、7.850、1.224 electrons/eV,其余峰值s與p分態(tài)密度近似等比降低. 態(tài)密度曲線(xiàn)向低能區(qū)移動(dòng)與態(tài)密度峰值降低說(shuō)明吸附后CO2的電子更多的占據(jù)低能量的能態(tài). 此外,對(duì)比不同吸附質(zhì)的態(tài)密度圖可知,吸附質(zhì)從CH4、H2O到CO2,態(tài)密度分布區(qū)間逐漸增大,CO2在能量更低的區(qū)域具有態(tài)密度分布,與吸附能大小關(guān)系相對(duì)應(yīng),雖然三種吸附質(zhì)在β-SiO2的吸附為物理吸附,但CO2吸附能更小,更易優(yōu)先吸附. 基于密度泛函理論第一性原理計(jì)算方法研究了CH4、H2O、CO2、β-SiO2(100)面以及吸附質(zhì)在β-SiO2(100)面吸附的性質(zhì),對(duì)比吸附質(zhì)在β-SiO2(100)面上高對(duì)稱(chēng)位的吸附能和態(tài)密度等特性,得到以下主要結(jié)論及認(rèn)識(shí): (1)CH4、H2O和CO2在β-SiO2(100)面的吸附能分布在-0.2 eV~ -0.1eV區(qū)間內(nèi),均大于-0.62 eV且小于0 eV,為物理吸附;最小吸附能大小依次為:CH4> H2O> CO2,即,CO2的吸附能力最強(qiáng),H2O次之,CH4最弱;各吸附質(zhì)的吸附能變化范圍均非常狹窄,表明不同吸附位的吸附影響較小. (2)各吸附質(zhì)在最穩(wěn)定吸附位與最不穩(wěn)定位的物理結(jié)構(gòu)發(fā)生了不同程度的鍵長(zhǎng)鍵角變化,但其鍵長(zhǎng)鍵角變化均小于1%,吸附能最大對(duì)應(yīng)的吸附位鍵角絕對(duì)變化率均大于吸附能最小對(duì)應(yīng)的吸附位,吸附質(zhì)的物理結(jié)構(gòu)變化微弱表明其所受作用力微弱. (3)各吸附質(zhì)處于最穩(wěn)定吸附位時(shí)基底的態(tài)密度基本重合,表明各吸附質(zhì)與β-SiO2表面相互作用相似且差異較小;CH4、H2O、CO2的態(tài)密度曲線(xiàn)均向低能量區(qū)偏移且峰值出現(xiàn)不同程度降低,吸附后電子更多地占據(jù)低能量的能態(tài),吸附后的結(jié)構(gòu)較吸附前更穩(wěn)定,且CO2在能量更低的區(qū)域具有態(tài)密度分布,更易優(yōu)先吸附. 通過(guò)吸附能、物理結(jié)構(gòu)與態(tài)密度研究從密度泛函理論的角度說(shuō)明了CH4、H2O、CO2在β-SiO2表面吸附作用的量子力學(xué)機(jī)理,對(duì)應(yīng)的宏觀(guān)表現(xiàn)即頁(yè)巖儲(chǔ)層主要礦物石英中含有一定量的吸附氣,且可通過(guò)注入CO2開(kāi)采CH4,以上研究?jī)?nèi)容對(duì)揭示CH4、H2O、CO2在石英含量高的頁(yè)巖中吸附機(jī)理具有重要意義.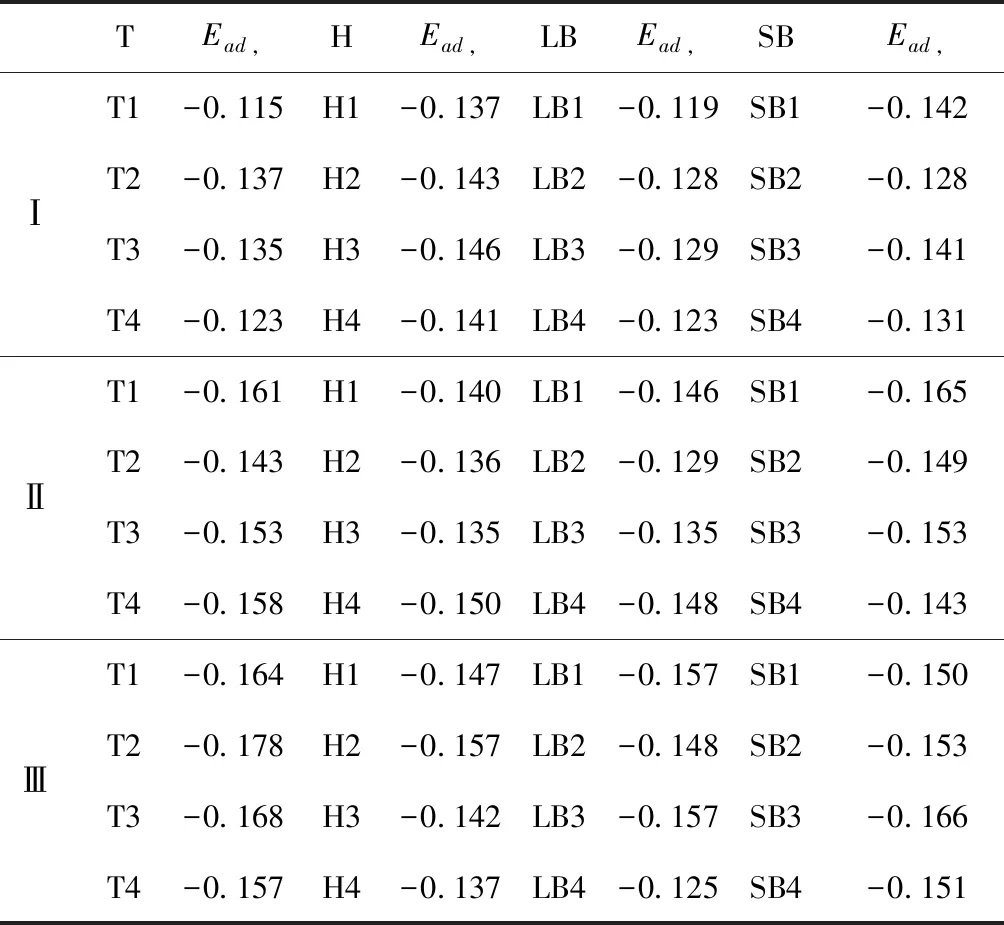
3.2 結(jié)構(gòu)分析
3.3 態(tài)密度分析


4 結(jié) 論
猜你喜歡
體育科技文獻(xiàn)通報(bào)(2022年3期)2022-05-23 13:46:54
天津外國(guó)語(yǔ)大學(xué)學(xué)報(bào)(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機(jī)設(shè)計(jì)與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車(chē)工程學(xué)報(bào)(2017年2期)2017-07-05 08:13:02
國(guó)際商務(wù)財(cái)會(huì)(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
