苦參轉(zhuǎn)錄組SSR位點及基因功能注釋分析
2019-09-17 07:32:25張寧尹美強譚青青
江蘇農(nóng)業(yè)科學(xué) 2019年7期
張寧 尹美強 譚青青
摘要:分析苦參轉(zhuǎn)錄組中的簡單重復(fù)序列(SSR)位點信息,為開發(fā)分子標(biāo)記奠定基礎(chǔ)。利用Fastqc軟件對苦參轉(zhuǎn)錄組測序的原始讀長(reads)進行質(zhì)量評估,再用Trimmomatic軟件對reads質(zhì)量較差的堿基進行過濾,利用Trinity軟件對Trimmomatic處理后的reads進行序列組裝,之后使用基因組裝完整性評估(BUSCO)軟件對轉(zhuǎn)錄組組裝的序列進行質(zhì)量評估,并分析組裝的conting序列的開放閱讀框(open reading frame,簡稱ORF);利用MicroSAtellite(MISA)軟件對無冗余獨立基因(unigene)進行SSR搜索。利用Trinity軟件最終篩選得到23074條ORF信息;使用MISA軟件從unigenes序列中發(fā)現(xiàn)8 798個SSR位點,分布于7 339條unigene中,總體上unigenes序列中SSR占比為2.16%,SSR位點平均間隔是5.28 bp,其中占比最高的是單核苷重復(fù)基序,為50.53%;其次是出現(xiàn)頻率分別為22.28%、24.73% 的二、三核苷酸。苦參轉(zhuǎn)錄組中SSR類型眾多,出現(xiàn)頻率高,在后續(xù)的苦參遺傳性狀分析,及次生代謝(苦參堿和黃酮等次生代謝產(chǎn)物)途徑等相關(guān)基因定位等方面具有很好的應(yīng)用潛力。
關(guān)鍵詞:苦參;轉(zhuǎn)錄組;SSR;位點信息;基因功能;分子標(biāo)記
中圖分類號: R285 ?文獻標(biāo)志碼: A ?文章編號:1002-1302(2019)07-0041-04
苦參(Sophora flavescens Ait.)是豆科槐屬植物,以其干燥根入藥,味苦,性寒,具有清熱除燥濕、殺蟲和利尿等藥效。其主要藥用成分是生物堿類和黃酮類化合物,已從苦參中分離出生物堿類39個,黃酮類122個成分[1]。苦參主產(chǎn)于山西、陜西、河南、河北等地,在醫(yī)學(xué)臨床、農(nóng)業(yè)、畜牧業(yè)和日用品等中有廣泛的應(yīng)用[2]。氣候的變化和人為過度的采挖造成野生苦參資源數(shù)量急劇減少[3]。因此,保護和利用好野生苦參資源是當(dāng)務(wù)之急,勢在必行。
分子標(biāo)記開發(fā)可對制定合理有效的種質(zhì)資源保護策略提供科學(xué)依據(jù),但目前還缺乏能夠應(yīng)用于苦參種質(zhì)鑒定、遺傳圖譜構(gòu)建、功能基因定位等研究的簡便、高效、穩(wěn)定且具有種屬特異性的分子標(biāo)記體系。簡單重復(fù)序列(simple sequence repeat,簡稱SSR)是由核苷酸構(gòu)成的重復(fù)序列,在真核生物和原核生物基因中都有存在。SSR 位點標(biāo)記具有在生物中分布廣泛、重復(fù)類型多樣、出現(xiàn)頻度高等特點[4],主要應(yīng)用于分子育種優(yōu)良基因定位、生物多樣性分析、遺傳圖譜繪制、突變體單核苷酸多態(tài)性(single nucleotide polymorphism,簡稱SNP)位點分析輔助等。傳統(tǒng)尋找基因組中SSR標(biāo)記的方法存在位點開發(fā)成本高、步驟較多、操作繁瑣等問題[5]。轉(zhuǎn)錄組SSR位點開發(fā)具有方便快捷、效率高等特點,且成本低廉。SSR開發(fā)引物能夠直接快速地定位基因信息。隨著苦參研究的深入,目前還未發(fā)現(xiàn)有關(guān)苦參轉(zhuǎn)錄組SSR開發(fā)的報道。本研究通過分析苦參轉(zhuǎn)錄組中的SSR位點信息,為苦參遺傳性狀分析、次生代謝(苦參堿和黃酮等次生代謝產(chǎn)物)途徑、分子標(biāo)記輔助育種及苦參遺傳多樣性研究提供依據(jù)和參考。
1 材料與方法
1.1 轉(zhuǎn)錄組數(shù)據(jù)來源
從NCBI(美國國家生物技術(shù)中心)數(shù)據(jù)共享平臺獲得苦參轉(zhuǎn)錄組數(shù)據(jù),從SRA(Sequence Read Archive)數(shù)據(jù)庫(https://www.ncbi.nlm.nih.gov/sra/)獲得苦參葉片RNA-Seq原始測序數(shù)據(jù),下載編號是SAMD00029896,使用Illumina HiSeq1000對苦參組織進行建庫測序,原始數(shù)據(jù)reads為 90 bp,采取雙端(paired-end sequencing)測序,獲得1.3 GB轉(zhuǎn)錄組數(shù)據(jù),下載網(wǎng)址是ftp://ftp.ncbi.nlm.nih.gov中的DRR031281[6]。
1.2 轉(zhuǎn)錄組的從頭組裝
首先通過Sratoolkit.2.8.2-1將sra格式轉(zhuǎn)錄組原始數(shù)據(jù)轉(zhuǎn)換為fastq格式[7];使用Fastqc軟件進行轉(zhuǎn)錄組原始數(shù)據(jù)質(zhì)量評估,然后,利用Trimmomatic軟件對fastq格式的序列進行低質(zhì)量去除,leading頭部去掉質(zhì)量低于3的堿基,trailing尾部過濾掉質(zhì)量低于3的堿基,每4個堿基是一個閱讀框,4個連續(xù)堿基的平均質(zhì)量低于15的過濾掉,reads中最小長度小于40序列的過濾掉 [8];隨后,對高質(zhì)量reads采用Trinity 軟件進行從頭(de novo)組裝[9],最短contig 長度設(shè)置為200 bp(參數(shù)為默認(rèn)參數(shù))。篩選每個基因最長的轉(zhuǎn)錄本作為unigene,最后組裝得到苦參轉(zhuǎn)錄組的全部轉(zhuǎn)錄本(包含可變剪切)。
1.3 苦參轉(zhuǎn)錄組數(shù)據(jù)組裝完整性評估
選取由Trinity軟件組裝的序列,使用BUSCO V 2.0.1軟件進行苦參葉片轉(zhuǎn)錄組數(shù)據(jù)完整性評價[10]。BUSCO V 2.0.1 軟件依據(jù) Ortho DB 數(shù)據(jù)庫,組成了幾個大的進化分支單拷貝基因集,將轉(zhuǎn)錄本reads拼接結(jié)果與該基因集數(shù)據(jù)進行比較(基因集直接使用 HMMER3與參考數(shù)據(jù)庫比對),依據(jù)比對上的比例、完整性評估拼接結(jié)果的準(zhǔn)確性和完整性。
1.4 ORF預(yù)測
使用Trinity軟件中的TransDecoder LongOrfs工具對unigene進行開放閱讀框(open reading frame,簡稱ORF)預(yù)測,篩選大于100個氨基酸的ORF序列,獲得最佳的ORF區(qū)域,使用Pfam (http://pfam.xfam.org/)和UniProt(http://www.uniprot.org)數(shù)據(jù)庫對預(yù)測結(jié)果進行校正,將比對結(jié)果保留到Pfam和UniProt數(shù)據(jù)庫的蛋白質(zhì)序列中[11]。
1.5 SSR位點搜索
使用MISA軟件[12]對苦參轉(zhuǎn)錄組數(shù)據(jù)unigene的SSR位點進行定位搜索,查詢定位規(guī)則是三堿基、四堿基、五堿基和六堿基重復(fù)至少5次,二堿基重復(fù)不得少于6次,2個SSR位點之間不足100bp則視為復(fù)合型SSR。
1.6 含SSR序列的基因功能注釋及生物堿基因挖掘
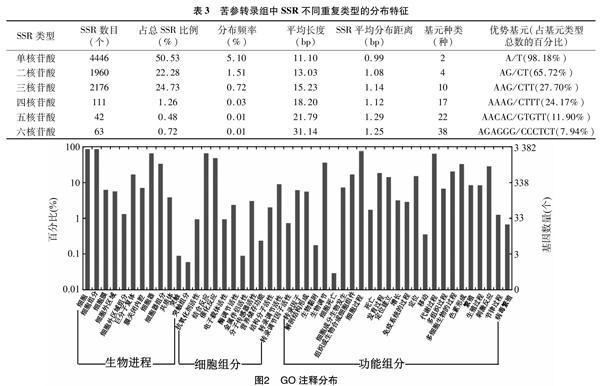
通過diamond blastx和diamond blastp分別將苦參中含SSR的8248條unigene序列與uniprot_sprot、Pfam和eggnog、Kegg、基因本體論(gene ontology,簡稱GO)等數(shù)據(jù)庫進行比對,比對參數(shù)e值<10-5,然后利用WEGO(http://wego.genomics.org.cn/)在線分析工具進行GO功能分類統(tǒng)計,分析含有SSR unigene的功能分布特征;通過與GO庫進行比對后,得到的unigene注釋結(jié)果按照GO數(shù)據(jù)庫的23個類別進行分類統(tǒng)計。通過對WEGO注釋結(jié)果(3個大類)23個子類更深入分析挖掘苦參堿相關(guān)基因,為進一步研究奠定基礎(chǔ)。
2 結(jié)果與分析
2.1 苦參轉(zhuǎn)錄組de novo 組裝
從NCBI數(shù)據(jù)庫下載得到的苦參轉(zhuǎn)錄組測序(RNA-Seq)數(shù)據(jù)中共包含14 636 096個雙端測序 reads,通過Trimmomatic軟件過濾得到14 578 802 個高質(zhì)量 reads。轉(zhuǎn)錄組 de novo組裝獲得53 179個長度大于200 bp的contigs,拼接獲得的長序列(contigs)平均長度為813 bp,最長的 contig為22 546 bp,N50為1 464 bp;篩選每個基因中最長的轉(zhuǎn)錄本,共得到54 221條unigenes,平均長度為715.87 bp,最長的unigene 為12 122 bp,N50為1 464 bp(表1)。采用TransDecoder軟件中LongOrfs功能進行ORF預(yù)測,篩選獲得大于100個氨基酸的ORF有29 226個contigs;通過UniProt蛋白質(zhì)數(shù)據(jù)庫比對獲得15 242條蛋白質(zhì)序列,Pfam數(shù)據(jù)庫比對獲得126 429條蛋白質(zhì)序列;使用TransDecoder最終篩選得到23 074條ORF信息。
contigs 和unigenes的鳥嘌呤(G)和胞嘧啶(C)占比都是44. 8%。從序列長度分布看,序列長度分布在1 000~2 899 bp 的序列大約有19.3%,≥2 900 bp的序列只有0.2%,600~999 bp的序列大約有13.6%,700bp 以下占71.4%(圖1)。
2.2 轉(zhuǎn)錄組數(shù)據(jù)完整性評估
對轉(zhuǎn)錄組數(shù)據(jù)進行評估、測序、組裝得到的轉(zhuǎn)錄序列覆蓋所有可能的轉(zhuǎn)錄本。評估轉(zhuǎn)錄組數(shù)據(jù)的大小和完整性。依據(jù)植物直系同源基因數(shù)據(jù)集對苦參的轉(zhuǎn)錄組數(shù)據(jù)完整性進行評估,由表2可知,在由苦參轉(zhuǎn)錄組序列與植物基因組匹配獲得的1440個植物單拷貝直系同源基因中,完全匹配到的直系同源基因( complete)有1000個,占總BUSCO的69.4%,部分片段匹配對應(yīng)到的單拷貝直系同源基因( fragment)有171個,占總BUSCO的11.9%;沒有匹配對應(yīng)到的植物單拷貝直系同源基因(missing)有269個,占總BUSCO的18.7%,完全匹配到的單拷貝直系同源基因(complete)有973個,占總BUSCO的67.6%,完全匹配到的多拷貝直系同源基因(complete)有27個,占總BUSCO的1.9%。
2.4 轉(zhuǎn)錄組中SSR 位點的分布特點
使用 Trinity軟件組裝得到54 221條unigenes,堿基數(shù)為 38 815 308 bp,平均每條unigene長度為715.87 bp;使用 MISA軟件搜索得到8 798個SSR位點,存在于7 339條unigenes轉(zhuǎn)錄組序列中,包括多個 SSR位點的 unigenes序列有1 173條(包含復(fù)合 SSR為551個)占SSR unigenes序列總數(shù)的13.33%。總體上unigenes序列中SSR占比為2.16%,SSR位點平均間隔距離是4 411 bp。其中占比最高的是單核苷重復(fù)基序,占總SSR 的50.53%;其次是出現(xiàn)頻率分別為22.28%、24.73% 的二、三核苷酸。SSR最短平均分布距離是0.99 bp的單核苷酸重復(fù)類型,平均分布距離最長的是1.29 bp的五核苷酸重復(fù)類型。
苦參轉(zhuǎn)錄組不同重復(fù)類型的SSR位點都有多種基元,在考慮堿基互補且包含復(fù)合重復(fù)基元的情況下,重復(fù)類型合計93種,其中六核苷酸38種,五核苷酸22種,四核苷酸類型17種,在篩選的 SSR中單核酸重復(fù)優(yōu)勢基元為A/T,占比最高,為總基元類型的98.18%,其次是二核苷酸重復(fù)類型優(yōu)勢類型基元AG/CT,為65.72%。三核苷酸重復(fù)類型的優(yōu)勢基元是AAG/CTT,占比27.70%;四、五、六核苷酸重復(fù)類型的優(yōu)勢基元分別是AAAG/CTTT、AACAC/GTGTT、AGAGGG/CCCTCT,所占的比例分別是24.17%、11.90%、7.94%(表3)。
2.5 轉(zhuǎn)錄組SSR 基序重復(fù)類型和頻率特征
不同重復(fù)類型苦參轉(zhuǎn)錄組SSR位點分布存在差異(表4)。單核苷酸重復(fù)類型設(shè)置重復(fù)數(shù)≥15次作為SSR位點的識別條件,因此在表中未分析單核苷酸類型。除單核苷酸外,各重復(fù)類型重復(fù)數(shù)在5~11次之間,隨重復(fù)次數(shù)的逐漸增加,頻率逐步降低。除單核苷酸外,5~7 次是主要集中次數(shù),占SSR類型總數(shù)的大多數(shù)。
2.6 含SSR序列的基因功能注釋及生物堿基因挖掘
為了解含有SSR序列苦參轉(zhuǎn)錄組序列的基因功能,本研究通過與公共蛋白數(shù)據(jù)庫進行比對,得到含有SSR序列的分類信息和功能注釋。結(jié)果發(fā)現(xiàn),uniprot_sprot、Pfam、eggnog、Kegg、GO分別注釋到3 094、3 162、3 061、3 138、3 467個基因。
GO注釋將基因功能分為生物進程(biological process)、細(xì)胞組分(cellular component)、功能組分(molecular function)大類,其下又分了很多子類,從不同角度對基因的功能進行分類注釋,各類間互相關(guān)聯(lián)。GO注釋可以全面描述苦參中SSR基因和基因產(chǎn)物的屬性。將搜索到含有SSR的unigene序列使用blastx比對到蛋白數(shù)據(jù)庫,取比對分值最高的為序列注釋信息。細(xì)胞組分注釋10312條,生物進程注釋11 200條,功能組分注釋4 376條。將含有SSR序列的3 467條unigene編號后與其對應(yīng)的GO分類號一起導(dǎo)入到GO分類圖形顯示在線分析工具WEGO 軟件中,得到其基因功能分布(圖2)。結(jié)果表明,在3 467條unigene序列中注釋信息獲得23 483個功能注釋,平均1條unigene有6.77個GO注釋。
苦參主要藥用成分是苦參堿和黃酮類物質(zhì),通過對含有SSR位點的序列進行GO注釋數(shù)據(jù)挖掘,獲得7個生物堿代謝途徑相關(guān)基因,2個黃酮類生物合成過程相關(guān)基因。
3 討論
苦參轉(zhuǎn)錄組 de novo組裝獲得51 606 個長度大于200 bp的contigs,使用uniprot和Pfam蛋白質(zhì)數(shù)據(jù)庫進行ORF比對校正,uniprot比對上15 242條蛋白質(zhì)序列,Pfam數(shù)據(jù)庫校比對上 126 429 條蛋白質(zhì)序列,TransDecoder最終篩選得到 23 074條ORF信息,unigenes序列長度在700 bp 以下的序列
數(shù)大約占總序列數(shù)的70%。BUSCO對轉(zhuǎn)錄組組裝結(jié)果:C占比為69.5%,S占比為67.6%,D占比為1.9%,F(xiàn)占比為11.9%,M占比為18.6%,總BUSCOs數(shù)目為1 440條。
苦參轉(zhuǎn)錄組序列通過MISA搜索到8 798個SSR位點,SSR位點的unigenes序列在苦參轉(zhuǎn)組序列中SSR位點占比為2.16%,平均分布距離4 411 bp出現(xiàn)1個SSR。與其他藥用植物比較,高于黨參的0.022%[13],低于丹參的0.047%[14],高于西洋參的0.013 3%[15]和人參的0.017 2%[16];與豆科模式植物大豆相比,高于大豆的0.013 5%[17]。表明苦參的SSR位點數(shù)量較為豐富。通過對含有SSR位點序列的注釋進一步分析獲得苦參生物堿相關(guān)代謝基因,為后續(xù)相關(guān)研究提供參考。
本研究結(jié)果為苦參轉(zhuǎn)錄組數(shù)據(jù)中的SSR位點分析提供依據(jù)。本研究對轉(zhuǎn)錄組序列進行了ORF預(yù)測,反映了基因組中基因的編碼區(qū)域,可進一步確定基因位置,省去了SSR引物設(shè)計開發(fā)過程中的克隆和測序步驟,充分利用了生物信息數(shù)據(jù)庫現(xiàn)有測序數(shù)據(jù),降低了開發(fā)成本。同時也明確了苦參SSR位點的基本特點,為進一步開發(fā)設(shè)計新的苦參功能基因SSR 標(biāo)記奠定了基礎(chǔ)。苦參中SSR對于苦參基因功能資源的開發(fā)利用、遺傳資源評估、豐富的分子標(biāo)記、種質(zhì)資源改良和比較基因組學(xué)研究都具有重要的價值。
參考文獻:
[1]國家藥典委員會. 中華人民共和國藥典[M]. 北京:化學(xué)工業(yè)出版社,2015.
[2]張貴君. 精編中草藥彩色圖譜[M]. 北京:中國醫(yī)藥科技出版社,2016.
[3]張 翅. 苦參莖葉中化學(xué)成分的研究[D]. 天津:天津中醫(yī)藥大學(xué),2013.
[4]段永紅,渠云芳,王長彪,等. 藥用植物苦參SSR-PCR體系的優(yōu)化與驗證[J]. 中國農(nóng)業(yè)大學(xué)學(xué)報,2014,19(5):95-100.
[5]He J Y,Zhu S,Komatsu K,et al. Genetic polymorphism of medicinally-used Codonopsis species in an internal transcribed spacer sequence of nuclear ribosomal DNA and its application to authenticate Codonopsis Radix[J]. Journal of Natural Medicines,2014,68(1):112-124.
[6]Han R,Takahashi H,Nakamura M,et al. Transcriptome analysis of nine tissues to discover genes involved in the biosynthesis of active ingredients in Sophora flavescens[J]. Biological and Pharmaceutical Bulletin,2015,38(6):876-883.
[7]Staff S. Using the SRA Toolkit to convert .sra files into other formats[EB/OL]. (2015-08-22)[2017-12-06]. http://www.ncbi.nlm.nih.gov/books/NBK158900/.
[8]Bonnal R J P,Ranzani V,Arrigoni A,et al. De novo transcriptome profiling of highly purified human lymphocytes primary cells[J]. Scientific Data,2015,2:150051.
[9]Grabherr M G,Haas B J,Yassour M,et al. Trinity:reconstructing a full-length transcriptome without a genome from RNA-Seq data[J]. Nature Biotechnology,2011,29(7):644-652.
[10]王 林. 白色鏈霉菌和白背飛虱的基因組學(xué)研究[D]. 合肥:中國科學(xué)技術(shù)大學(xué),2017.`
[11]舒江平,劉 莉,沈 慧,等. 基于系統(tǒng)基因組學(xué)分析揭示早期陸生植物的復(fù)雜網(wǎng)狀進化關(guān)系[J]. 生物多樣性,2017,25(6):675-682.
[12]王 希,陳 麗,趙春雷. 利用MISA工具對不同類型序列進行SSR標(biāo)記位點挖掘的探討[J]. 中國農(nóng)學(xué)通報,2016,32(10):150-156.
[13]王 東,曹玲亞,高建平. 黨參轉(zhuǎn)錄組中SSR位點信息分析[J]. 中草藥,2014,45(16):2390-2394.
[14]鄧科君,張 勇,熊丙全,等. 藥用植物丹參EST-SSR標(biāo)記的鑒定[J]. 藥學(xué)學(xué)報,2009,44(10):1165-1172.
[15]楊維澤,金 航,趙振玲,等. 西洋參EST資源的SSR信息分析[J]. 西南農(nóng)業(yè)學(xué)報,2011,24(1):275-278.
[16]Li C F,Zhu Y J,Guo X,et al. Transcriptome analysis reveals ginsenosides biosynthetic genes,microRNAs and simple sequence repeats in Panax ginseng C. A. Meyer[J]. BMC Genomics,2013,14:245.
[17]Dreisigacker S,Zhang P,Warburton M L,et al. SSR and pedigree analyses of genetic diversity among CIMMYT wheat lines targeted to different megaenvironments[J]. Crop Science,2004,44(2):381-388. 么大軒,張 彬,劉松濤,等. 基于SNP和SSR對甜玉米種質(zhì)遺傳多樣性的評價[J]. 江蘇農(nóng)業(yè)科學(xué),2019,47(7):45-50.
