改性杏殼活性炭的制備、表征及其吸附乙烯性能
2019-09-28 02:01:30鄧叢靜馬歡歡周建斌
生物質化學工程 2019年5期
鄧叢靜, 馬歡歡, 周建斌*
(1.國家林業和草原局 林產工業規劃設計院, 北京 100010;2.南京林業大學 材料科學與工程學院, 江蘇 南京 210037)
乙烯是一種果蔬催熟劑,根據來源不同分為內源乙烯和外源乙烯,內源乙烯是植物內合成的,它能引起特定基因的表達,從而產生乙烯促進衰老的特有效應[1-3]。大部分果品、蔬菜在貯藏過程中,其鮮重的損失率和乙烯的濃度成正比[4],果蔬后熟一旦開始,自身乙烯的產生速率遠遠大于乙烯通過果肉果皮擴散到周圍環境中的擴散率,大大增加了腐爛速度[5]。目前,果蔬貯藏形式大都采用低溫冷藏,冷激處理可以延長果蔬的貯藏壽命,但即使在低溫環境中,只要有少量乙烯存在,果蔬的貯藏壽命還是會受到影響[5],如在0 ℃環境中貯藏獼猴桃,當乙烯體積分數為0.001%時,仍然會加速獼猴桃的軟化[6-7]。因此,清除貯藏環境中乙烯氣體、控制乙烯體積分數,有利于延長果蔬的保鮮期。清除環境中的乙烯是果蔬貯存采用的一個手段,常用的方法有通風換氣、氧化劑氧化法、催化氧化法、物理吸附法、乙烯脫除膜法等[8-9]。物理吸附法主要是采用吸附劑清除外源或內源乙烯,乙烯吸附劑有活性炭、分子篩、天然有機吸附劑、天然無機吸附劑等。活性炭是目前使用最廣泛的吸附劑,在吸附乙烯方面表現出明顯優勢。活性炭的孔結構和表面化學性質對其吸附性能有重要影響,改性處理可以有效提高活性炭的苯吸附性能[10-14]。本研究以氧化預處理活性炭為載體,研究了不同改性劑和改性條件對改性活性炭吸附乙烯性能的影響,以期為改性活性炭吸附乙烯的研究提供基礎數據。
1 實 驗
1.1 原料、試劑與儀器
雙氧水、硝酸銅、硝酸銀,均為分析純;杏殼活性炭(AC),承德華凈活性炭有限公司,粉碎至一定粒徑,備用;乙烯氣體(純度99.999%),南京特種氣體廠有限公司。
WH-201氣候箱,南京實驗儀器廠;1900型比表面積測定儀,意大利Sorptomatic公司;Autopore Ⅱ9220型壓汞儀,美國Micromeritiecs公司;Quanta 200型環境掃描電鏡(ESEM),荷蘭Philips-FEI公司;X-act型X射線能譜(EDS)儀,英國牛津INCA公司;Nicolet 170SFT型傅里葉變換紅外光譜(FT-IR)儀,美國Theme electron公司;VG ESCALAB MKⅡ型的X射線光電子能譜(XPS)儀,英國Vaccum Generator Scientific公司。
1.2 活性炭樣品的改性
1.2.1改性試劑的選擇
1.2.1.1硝酸銀改性活性炭 取10 g粒徑0.38~0.83 mm活性炭置于坩堝中加入10 mL一定質量分數的AgNO3溶液,采用浸漬法對活性炭進行改性處理。浸漬過程中,每15 min攪拌一次,浸漬6 h后,于120 ℃烘干至質量恒定,然后放入馬弗爐中,400 ℃條件下焙燒4 h,自然冷卻至室溫,即得硝酸銀改性活性炭(Ag-AC)。AgNO3溶液質量分數分別為1%、2%、3%、4%、5%和6%。
1.2.1.2硝酸銅改性活性炭 改性操作同1.2.1.1節,所用改性劑為Cu(NO3)2溶液,質量分數分別為1%、2%、3%、4%、5%和6%。所得產物為硝酸銅改性活性炭(Cu-AC)。
1.2.1.3雙氧水-硝酸銅改性活性炭 稱取100 g活性炭,加入100 mL質量分數15%的雙氧水,混合至無氣泡產生后,于80 ℃加熱1 h,趁熱過濾后用去離子水清洗至洗液為中性后,于120 ℃烘箱中烘干,即得雙氧水預處理活性炭,表示為H2O2-AC。然后按1.2.1.2操作,僅AC用H2O2-AC替換,所得產物為H2O2-Cu-AC。
1.2.2改性條件的確定
1.2.2.1活性炭粒徑 取4種不同粒徑活性炭(0.83~1、0.38~0.83、0.23~0.38和0.15~0.23 mm),采用15% H2O2進行氧化預處理,然后用改性試劑2%Cu(NO3)2對活性炭進行浸漬,浸漬6 h后,于120 ℃ 烘干,再放入馬弗爐中,400 ℃焙燒4 h,自然冷卻至室溫,然后在20 ℃恒溫條件下,吸附乙烯24 h,測量其對乙烯的吸附量,研究粒徑對活性炭吸附乙烯性能的影響。
1.2.2.2浸漬時間 選擇活性炭粒徑0.38~0.83 mm,采用15%H2O2和2%Cu(NO3)2分別對其進行氧化預處理和改性處理,分別浸漬 2、4、6、8 h后,其他條件同1.2.2.1節,研究浸漬時間對改性活性炭吸附乙烯性能的影響。
1.2.2.3焙燒時間 選擇活性炭粒徑0.38~0.83 mm,采用15%H2O2和2%Cu(NO3)2分別對其進行氧化預處理和改性處理,分別焙燒 2、4、6、8 h,其他條件同1.2.2.1節,研究焙燒時間對改性活性炭吸附乙烯性能的影響。
1.2.2.4焙燒溫度 采用15%H2O2和2%Cu(NO3)2分別對粒徑0.38~0.83 mm的活性炭進行氧化預處理和改性處理,分別在 200、400、600、800 ℃焙燒6 h,其他條件同1.2.2.1節,研究焙燒溫度對改性活性炭吸附乙烯性能的影響。
1.3 吸附乙烯實驗
稱取1 g (稱準至0.5 mg)干燥試樣,放入預先干燥和稱量過的稱量瓶中,試樣在稱量瓶底部搖晃至厚度均勻,打開稱量瓶瓶蓋,放入真空干燥器內,然后將干燥器抽真空至0.1 MPa,并保持10 min。將乙烯氣體導入真空干燥器(直徑D=30 cm,高H=40 cm)內,氣流速度緩慢以防試樣被氣流沖出稱量瓶,當真空干燥器內壓力恢復至常壓時,停止導入乙烯氣體,并將真空干燥器與裝有乙烯氣體的氣囊相連,保持整個吸附過程中真空干燥器內壓力恒定。將連有乙烯氣囊的真空干燥器放入20 ℃恒溫箱中,恒溫吸附24 h后,稱質量計算吸附量,同時做空白實驗。吸附量計算公式如下:
(1)
式中:Q—試樣的乙烯吸附量,g/g;m—稱量瓶的質量,g;m1—吸附前試樣加稱量瓶的質量,g;m2—吸附后試樣加稱量瓶的質量,g;m3—空白實驗質量增加量,g。
1.4 樣品表征
1.4.1ESEM-EDS表征 將干燥試樣在瑪瑙研缽中研磨至粒徑<0.071 mm,經壓片后,在環境掃描電鏡(ESEM)和X射線能譜(EDS)儀上進行測試。ESEM測試條件為表面噴金,工作電壓10~25 kV,樣品放大150~20 000倍。EDS測試條件為電壓20 kV,工作距離10.0 mm。
1.4.2比表面積和孔徑分布 取粒徑<0.071 mm的干燥試樣用比表面積測定儀(液氮)進行測定。
1.4.3紅外光譜(FT-IR) 表征 取粒徑<0.071 mm干燥試樣,溴化鉀壓片法制樣,經紅外光譜儀分析,掃描范圍為500~4000 cm-1。
1.4.4X射線光電子能譜(XPS)分析 取粒徑<0.071 mm干燥試樣,經壓片后,在光電子能譜儀上進行測試。測試條件為功率250 W,電壓12.5 kV,電流20 mA,光源Al-Kα。
2 結果與討論
2.1 改性試劑對改性活性炭吸附乙烯的影響
采用不同改性劑按1.2.1節方法制備不同改性活性炭,其在20 ℃恒溫吸附乙烯24 h的結果見圖1。

a.Ag-AC; b.Cu-AC; c.H2O2-Cu-AC
從圖1(a)可以看出,隨著改性劑硝酸銀質量分數的增加,活性炭對乙烯的吸附量先增加后降低。當硝酸銀質量分數為2%時,改性效果最佳,乙烯吸附量為0.089 g/g,比未改性的杏殼活性炭(AC)的吸附量(0.069 g/g)提高了28.99%,隨著改性試劑質量分數的繼續增加,改性活性炭對乙烯的吸附量逐漸減少。這是因為銀離子具有催化活性,可以活化C—H鍵,使其更容易與活性炭孔內較強的活性中心發生反應,從而達到吸附乙烯的效果。隨著改性試劑質量分數的增加,活性組分銀在活性炭孔隙內部沉積,堵塞了部分孔隙結構,使活性炭孔內氧化活性降低,導致乙烯吸附量降低。
從圖1(b)可以看出,活性炭對乙烯的吸附量隨著硝酸銅溶液質量分數的增加先升高后降低,當硝酸銅溶液質量分數為2%時,改性活性炭對乙烯的吸附效果最好,吸附量為0.132 g/g,比AC吸附量提高了91.30%。這是因為銅離子具有催化活性,可以活化C—H鍵,使其更容易與活性炭孔內較強的活性中心發生反應,從而達到吸附乙烯的效果。隨著改性試劑質量分數的增加,活性組分銅離子在活性炭孔隙內部沉積,堵塞了部分孔隙結構,使活性炭孔內氧化活性降低,導致乙烯吸附量降低。
對比圖1(a)和(b),可以看出硝酸銅改性活性炭比硝酸銀改性活性炭對乙烯的吸附效果好,可能是由于C2H4以側基與銅離子發生絡合作用[14]。
從圖1(c)可以看出,H2O2-Cu-AC對乙烯的吸附量隨著硝酸銅質量分數的增加先增加后降低,Cu(NO3)2為2%時對乙烯的吸附效果最好,吸附量為0.163 g/g,比AC提高了136.23%。
對比圖1(b)和(c)可以看出,硝酸銅溶液質量分數相同時H2O2-Cu-AC對乙烯的吸附量明顯高于Cu-AC,這是因為活性炭經雙氧水處理后,其表面酸性官能團增加,有利于銅離子分散。因此,后續實驗的選擇15%H2O2和2%Cu(NO3)2為預處理劑和改性劑。

表1 活性炭粒徑對乙烯吸附量的影響
2.2 改性條件對改性活性炭吸附乙烯的影響
2.2.1活性炭粒徑 按照1.2.2.1節中的方法制備改性活性炭H2O2-Cu-AC,其吸附乙烯結果如表1所示。
由表1可知,改性活性炭對乙烯的吸附量受活性炭粒徑的影響較大,在試驗范圍內,粒徑為0.38~0.83 mm的活性炭,改性后對乙烯的吸附效果最好,吸附量為0.163 g/g。改性條件相同的情況下,對于不同粒徑的活性炭,粒徑大的則由于改性劑與碳的接觸面積小,可能會發生外部燒失,而內部未完全活化,導致粒徑大于0.83 mm 的活性炭改性后對乙烯的吸附量小;對于粒度小的,隨著活性炭粒度的進一步變小,在改性過程中可能會被過度燒失,導致乙烯吸附量降低。
2.2.2浸漬時間 不同浸漬時間制備的H2O2-Cu-AC吸附乙烯的結果見圖2(a),由圖可知不同浸漬時間對改性活性炭吸附乙烯性能的影響較小,浸漬6 h時對乙烯的吸附效果最好。
2.2.3焙燒時間 不同焙燒時間制備的H2O2-Cu-AC吸附乙烯的結果如圖2(b)所示。由圖可知,當焙燒時間為4 h時,改性活性炭對乙烯的吸附性能最好,焙燒時間過長或過短都不利于改性活性炭對乙烯氣體的吸附。
2.2.4焙燒溫度 不同焙燒溫度下制備的H2O2-Cu-AC,其吸附乙烯的結果如圖2(c)所示。由于焙燒溫度800 ℃時,活性炭燒失嚴重,未進行吸附試驗,故沒有相應吸附數據。由圖可知,焙燒溫度為 400 ℃時,改性活性炭對乙烯的吸附性能最好,吸附量為0.163 g/g。焙燒溫度過低時,焙燒負載不完全,達不到較好的吸附性能;焙燒溫度過高,活性炭上負載的金屬離子易被還原為金屬單質,不利于對乙烯的絡合吸附,且焙燒溫度過高,活性炭嚴重燒失,改性活性炭得率很低。
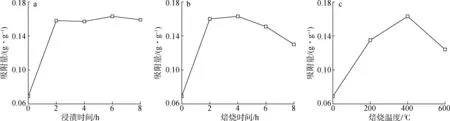
a.浸漬時間immersion time; b.焙燒時間roasting time; c.焙燒溫度roasting temperature
綜上,活性炭改性的較佳條件為15%雙氧水預處理、改性劑2%硝酸銅、活性炭粒徑0.38~0.83 mm、浸漬時間6 h、焙燒溫度400 ℃、焙燒時間4 h,此條件下制得H2O2-Cu-AC作后續研究。
2.3 改性活性炭H2O2-Cu-AC的表征
2.3.1ESEM-EDS分析 對較佳活化條件下制得的H2O2-Cu-AC進行ESEM-EDS分析,結果見圖3。

a.ESEM; b.EDS
從圖3可知,H2O2-Cu-AC中活性組分銅能分散在活性炭載體的表面和孔隙內部,說明硝酸銅的質量分數為2%時,銅在活性炭上的負載量較好,這與2.1節的實驗結果相同。
圖4為H2O2-Cu-AC的EDS面掃描示意圖,圖中白色小點表示元素分布情況,可以看出C元素是構成活性的主要元素,其分布集中,有團聚現象,凹凸不平,O元素主要分布在C元素的周圍,Cu元素的分布則相對較均勻。

a. H2O2-Cu-AC; b.C; c.O; d.Cu
2.3.2 比表面積和孔徑分布 活性炭的比表面積和孔徑分布是影響活性炭物理吸附的主要因素,對AC和H2O2-Cu-AC的比表面積和孔徑分布的測定結果如表2所示。
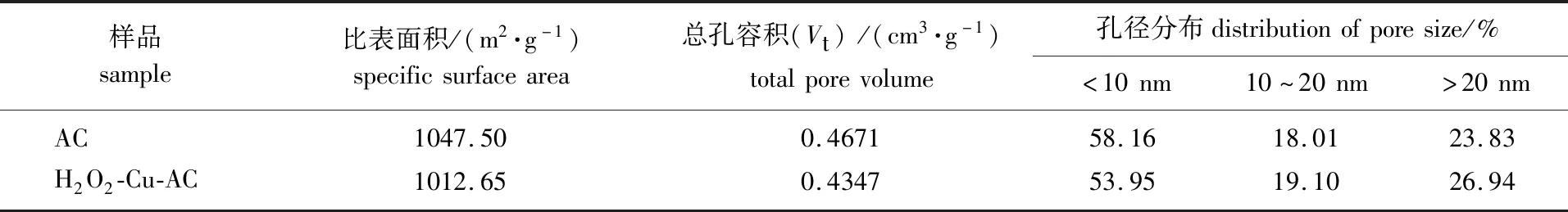
表2 活性炭的比表面積及孔徑分布
由表2可以看出,活性炭經15%H2O2和2%Cu(NO3)2處理后,其比表面積和總孔容積有所下降,表明改性劑負載到活性炭上堵塞了少部分孔隙,另外焙燒過程中少部分微孔被燒蝕,導致孔徑向較寬方向分布。

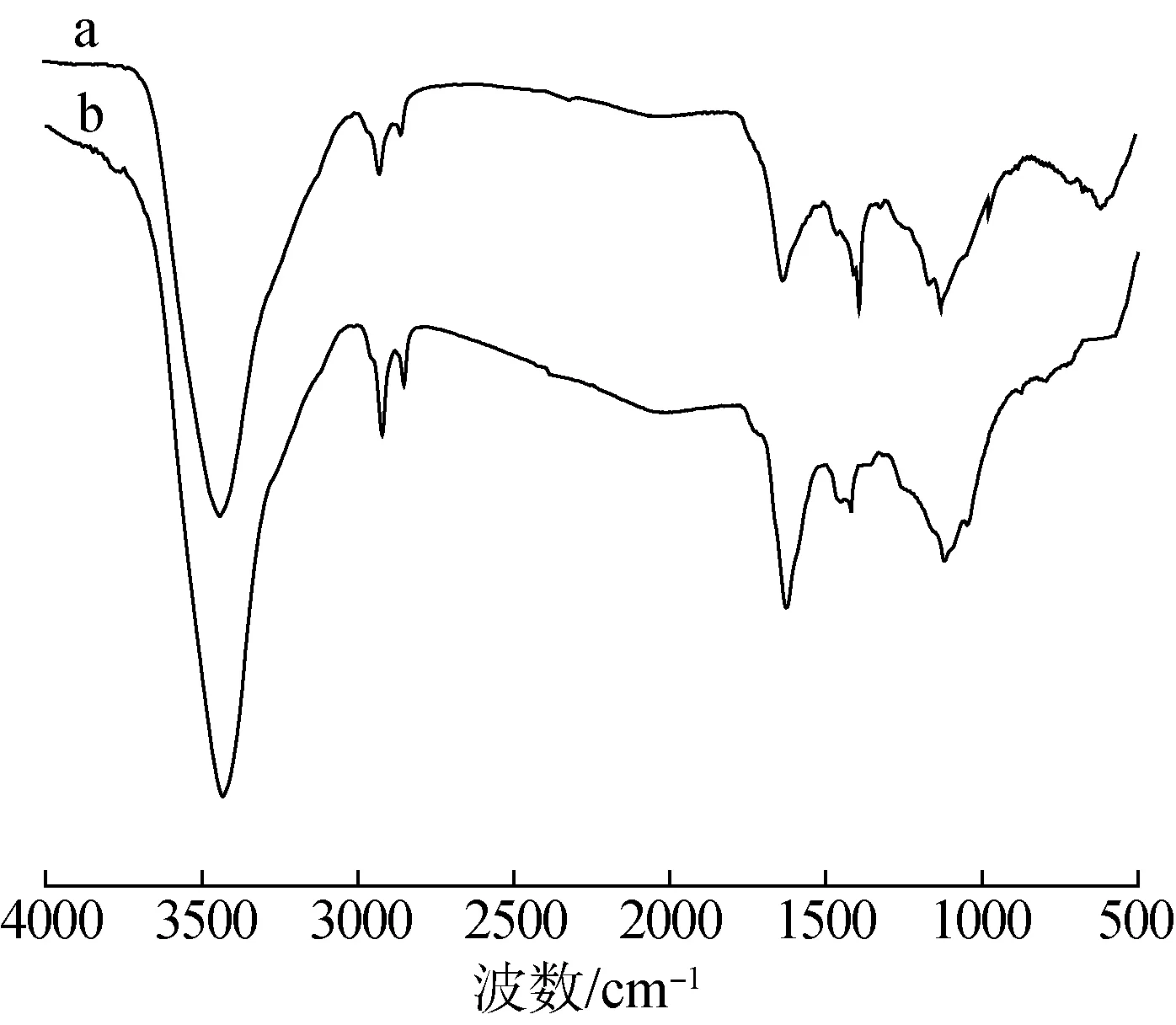
對比可知,H2O2-Cu-AC在1630~1389 cm-1處的吸收峰相對于AC明顯增強,1120 cm-1處的酚羥基吸收峰經過氧化處理后有所增強。結果表明H2O2-Cu-AC表面含氧官能團數量明顯增加。
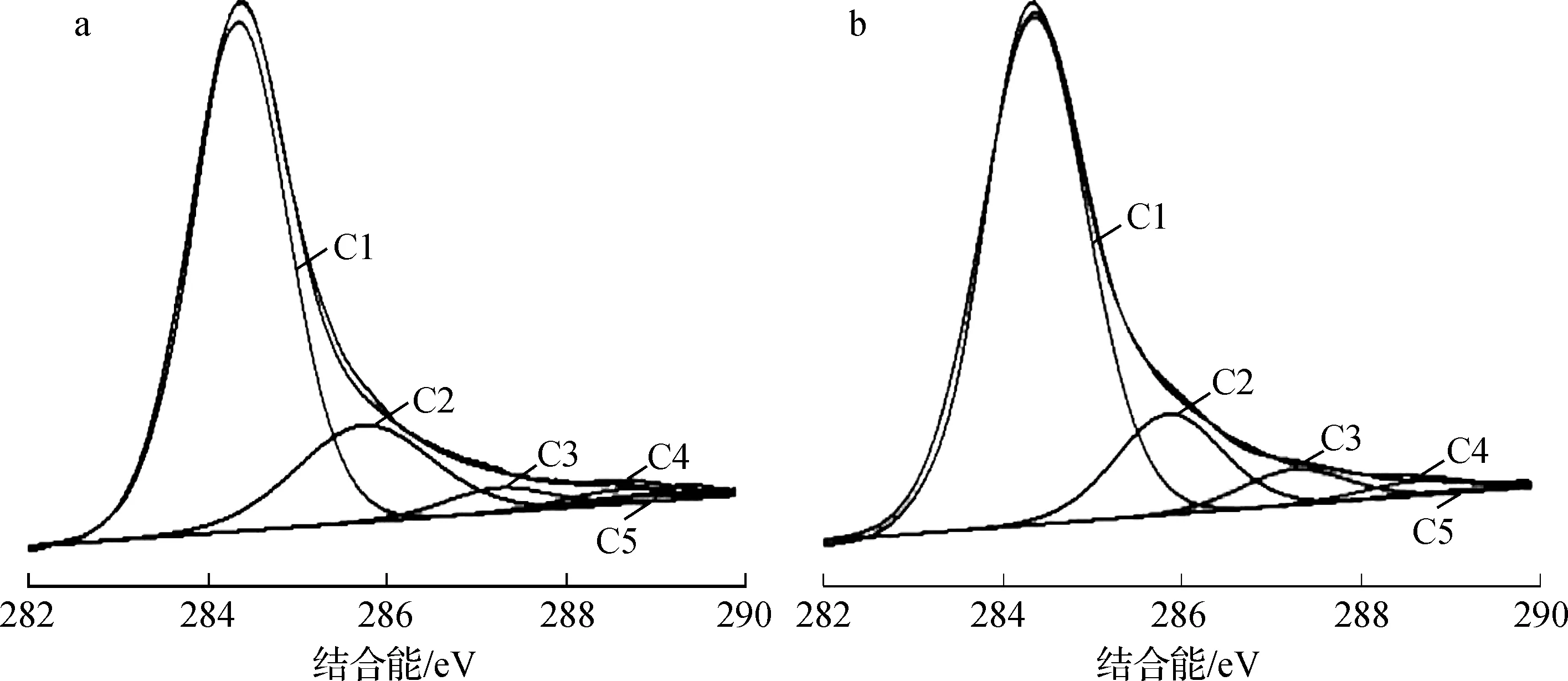
2.3.4 X射線光電子能譜分析 活性炭表面元素以及官能團的種類和含量的多少決定了活性炭表面的化學性質,進而影響活性炭對乙烯的吸附性能。H2O2-Cu-AC的XPS全掃描譜圖如圖6所示,AC作對照。
a.AC; b.H2O2-Cu-AC

圖6 改性活性炭的XPS圖
通過XPS寬掃描譜圖得出改性活性炭表面元素含量變化情況如表3。由表可以看出,改性前后活性炭的表面元素均以碳和氧為主,活性炭改性后,氧含量增加,O/C增加,說明改性后活性炭表面含氧官能團增加。銅和氮含量增加,主要是負載改性劑引起的。

表3 改性活性炭表面元素組成
a.AC; b.H2O2-Cu-AC
3 結 論
3.1對杏殼活性炭(AC)進行改性后用于吸附乙烯,考察了改性條件對改性活性炭吸附乙烯性能的影響,結果表明:用15%H2O2先氧化預處理,然后用2%Cu(NO3)2作改性劑時制得的改性活性炭吸附乙烯性能較好。活性炭粒度、焙燒時間和焙燒溫度對改性活性炭吸附乙烯性能的影響比較明顯,而浸漬時間的影響較小。在15%雙氧水氧化預處理、改性劑2%硝酸銅、活性炭粒度0.38~0.83 mm、浸漬時間6 h、焙燒溫度400 ℃、焙燒時間4 h的條件下制得改性活炭對乙烯吸附量為0.163 g/g,比未改性的活性炭(0.069 g/g)提高了136.23%。
3.2采用ESEM-EDS、FT-IR、XPS等手段對H2O2-Cu-AC進行表征。分析表明H2O2-Cu-AC中活性組分銅能相對較均勻地分散在活性炭載體的表面和孔隙內部;相較于AC,H2O2-Cu-AC的比表面積和總孔容積均有所下降,孔徑向較寬方向分布。改性劑引起了活性炭表面官能團的變化,其含氧官能團增加,C1、C3、C5降低,改性后C1峰面積由11 181增加到11 755,面積占比由77.468%降低到76.827%;C3峰面積由930降低到852,面積占比由6.684%降低到5.675%;C5峰面積由122降低到105,面積占比由0.844%降低到0.749%;C2、C4增加,改性后C2峰面積由1 950增加到2 329,面積占比由13.514% 增加到15.225%;C4峰面積由207增加到331,面積占比由1.490%增加到1.524%。
猜你喜歡
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中國塑料(2016年12期)2016-06-15 20:30:07
中國塑料(2016年5期)2016-04-16 05:25:36
汽車觀察(2016年3期)2016-02-28 13:16:26
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
