顱蓋骨腫物34例診治分析
2019-10-22 07:09:28呂健權瑜王舉波高李貴鞏守平
疑難病雜志 2019年10期
呂健,權瑜,王舉波,高李貴,鞏守平
顱蓋骨腫物并不少見,種類較多[1],其中以腫瘤居多,包括原發性腫瘤和繼發性腫瘤,僅根據臨床和影像學表現有時不易鑒別,但鮮有深入研究,文獻以個案報道居多。本文回顧性分析34例顱蓋骨腫物病例,探討其診斷、鑒別診斷和處理原則,以期提高對顱蓋骨腫物的認識。
1 臨床資料
1.1 一般資料 自2011年1月—2018年12月于西安交通大學第二附屬醫院神經外科收治34例顱蓋骨腫物病例,其中男13例,女21例;年齡1~76(39.15±2.07)歲;病程10 d~25年,平均(41.58±7.6)月。
1.2 臨床表現 均因“發現頭部腫物”就診,其中腫物緩慢(病程≥1年)進行性增大者9例(包括術后復發者1例);短期(病程<6個月)內出現并進行性增大者16例;發現多年、大小穩定但因外傷或無誘因在短期內明顯增大者4例;發現多年、大小穩定者4例;短期內偶然發現、大小穩定者1例;伴頭痛或局部疼痛者13例,伴肢體麻木、發熱、周圍性面癱、表面皮膚破潰、貧血各1例。觸診19例質地硬,5例質地較軟,10例質地中等。身體其他部位腫瘤手術史2例(腎細胞癌、直腸癌各1例)。
1.3 實驗室檢查 3例接受骨髓穿刺檢查,1例骨髓象表現為“增生性骨髓、巨核細胞易見、粒紅可見病態造血現象、網狀細胞易見”,1例表現為“中央骨髓中度抑制,外周骨髓輕度擴張”,1例診斷為“組織細胞增生癥”。
1.4 影像學檢查 均行顱腦CT檢查,表現為顱骨局部隆起或破壞,其中22例伴局部骨質破壞并接受MR檢查(平掃+增強)。多發2例,單發32例。最大徑1.5~6 cm,中位數3 cm。病變部位:左額骨4例,右額骨6例,雙側額骨多發1例;左頂骨7例,右頂骨5例,雙側頂骨多發1例,頂部中線2例;左顳骨3例,右顳骨2例(1例累及顱中窩底);枕骨3例。5例做全身ECT,其中1例發現雙側鎖骨異常放射性濃集,1例發現左側頂骨、左側第10肋椎關節、第1腰椎椎體、右側第5、第6前肋異常放射性濃集,1例發現顱骨、右肩關節、右肘關節異常放射性濃集,2例無異常。
1.5 最后診斷 1例經抗生素治愈而臨床診斷為感染性病灶,1例經影像學診斷為骨纖維異常增殖癥,1例因腎細胞癌病史及ECT發現多處骨轉移而診斷為顱骨轉移瘤,其余31例均經組織學確診,包括骨瘤12例(圖1),Langerhans組織細胞增生癥(Langerhans cell histiocytosis,LCH)5例(圖2),骨纖維異常增殖癥4例(圖2),腦膜瘤2例(間變性、內皮型各1例)(圖3),海綿狀血管瘤(圖3)、Rosai-Dorfman病(R-D病)(圖4)、轉移瘤(支氣管黏膜小細胞癌)(圖1)、非霍奇金B細胞淋巴瘤(non-Hodgkin's lymphoma,NHL)(圖1)、漿細胞瘤、多發性骨髓瘤、惡性纖維組織細胞瘤(圖1)和骨樣骨瘤各1例。11例初步診斷與最后診斷不符,占32.35%(11/34)。
1.6 治療與預后 首選手術治療。疑似感染性疾病者先行抗生素治療。惡性腫瘤或系統性疾病采取綜合治療。病變若僅累及外板,切除腫瘤及受累的外板和板障,保留內板;累及全層者切除全部受累顱骨,一期完成顱骨修補術。
1例多發性骨髓瘤和1例肺癌顱骨轉移接受化療,1例腎細胞癌顱骨轉移和1例惡性纖維組織細胞瘤放棄治療,1例感染性病灶經抗生素治愈(圖3),1例骨纖維異常增殖癥選擇臨床隨訪,其余28例均手術切除,其中局麻3例,全麻25例;次全切除2例(2/28,7.14%),全切除26例(26/28,92.86%)。6例術中見硬腦膜被侵襲,切除受侵襲的硬腦膜并用人工硬腦膜修補。一期顱骨修補術16例。2例遺留顱骨缺損未修補。術后并發癥包括皮下積液2例和切口延期愈合1例。
NHL和肺癌顱骨轉移患者均隨訪6個月,未見病變進展或復發;惡性纖維組織細胞瘤和腎細胞癌顱骨轉移患者失訪;其余30例隨訪6個月~5年,未見病變進展或復發。
2 典型病例
例1.女,30歲,以“發現左頂部進行性增大伴疼痛的腫物2月余”為主訴入院。查體可見左頂部軟組織腫物,質硬,無搏動,壓痛(+),不隨頭皮移動,表面皮膚色澤正常。X線顯示左頂骨內、外板類圓形溶骨性破壞。CT見左頂部軟組織腫物伴局部骨質缺如、骨邊緣不平整。MR顯示左頂部團塊呈T1等信號、T2混雜稍高信號,與周圍骨質界限不清,呈不均勻明顯強化,鄰近腦組織受壓。胸部X線見右側第5、第6肋骨局部膨大破壞。全身ECT見左側頂骨、左側第10肋椎關節、第1腰椎椎體、右側第5~6前肋異常放射性濃集。初步診斷為“嗜酸性肉芽腫”,骨髓細胞檢查報告“組織細胞增生癥”。全麻下開顱,見直徑4 cm的暗黃色魚肉樣腫物穿破顱骨全層,質脆,易出血,帽狀腱膜和硬腦膜均被侵襲,硬腦膜呈紫紅色,滲血多,腫物突破硬腦膜與腦表面粘連,腦表面被侵襲,徹底切除腫物及受侵襲的硬腦膜、顱骨。硬腦膜缺損和顱骨缺損分別用自體骨膜和鈦網修補。組織學診斷為LCH,Ki-67指數為20%,P53散在(+)。術后轉血液內科給予激素和化療。隨訪第18個月,ECT顯示左側髂骨近骶髂關節區又出現一處新的異常放射性濃集,其余病灶穩定。
例2.女,48歲,以“頭皮腫物切除術后10月,復發1月余”為主訴入院。10個月前,偶然發現右頂部頭皮腫物,在當地醫院切除但未做病理檢查。1個月前,發現腫物復發。查體可見右頂部中線旁粉紅色腫物,無壓痛及破潰。顱骨切線位X線見右頂骨缺損。MR見右頂部中線旁1個不規則形結節狀顱內外溝通性腫物,均勻明顯強化,伴腦膜尾征和腦水腫。MRV見上矢狀竇受累,但未見充盈缺損。初步診斷為“腦膜瘤”。術中見右頂骨中線旁有2處骨缺損,腫物呈灰紅色,質韌,血供豐富,穿破顱骨和硬腦膜,與上矢狀竇粘連緊密,腦表面受壓并被侵襲。徹底切除腫物及受累的硬腦膜和顱骨。硬腦膜缺損和顱骨缺損分別用自體骨膜和鈦網修補。組織學診斷為“R-D病”。骨髓細胞檢查顯示增生性骨髓、巨核細胞易見、粒紅可見病態造血現象、網狀細胞易見。術后口服潑尼松1個月。隨訪12個月,未見復發。
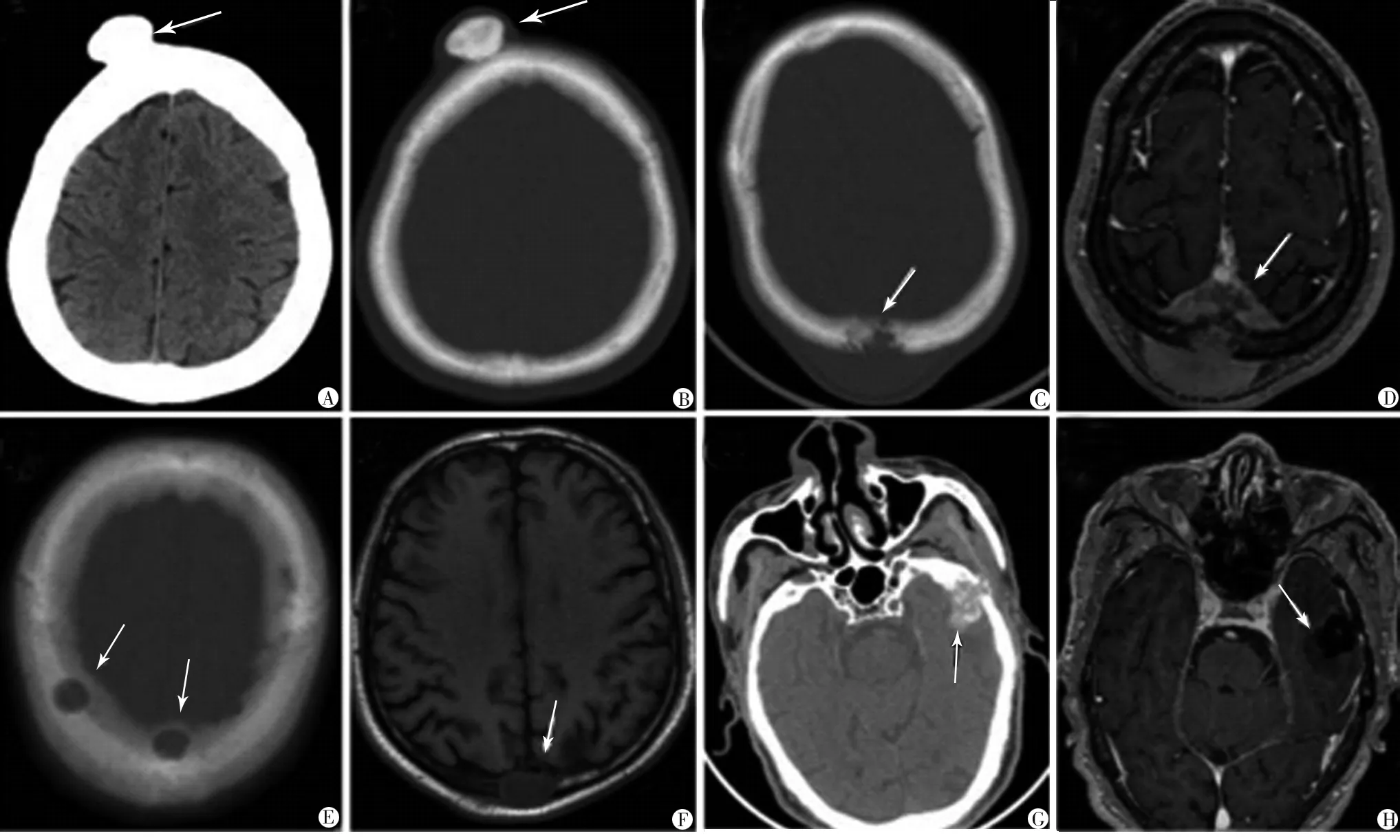
注:A、B.骨瘤,CT及其骨窗位可見起源于顱骨外板的廣基規則高密度影;C、D.非霍奇金B細胞淋巴瘤,CT和MR均可見顱頂中線頭皮下腫物伴局部骨質破壞,上矢狀竇受累;E、F.肺癌顱骨轉移,CT可見雙側頂骨多發類圓形低密度灶,MR可見頂部中線軟組織腫物;G、H.惡性纖維組織細胞瘤,CT可見骨破壞及軟組織影,MR增強可見病變內有壞死
圖1 骨瘤、轉移瘤、淋巴瘤、惡性纖維組織細胞瘤的影像學表現
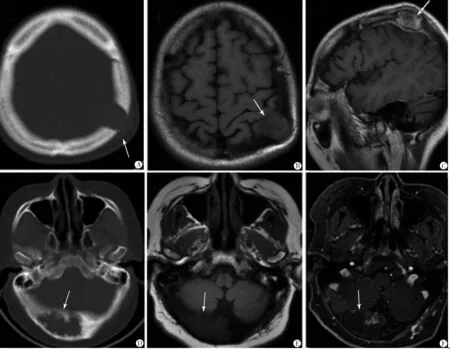
注:A~C.LCH,顱腦CT可見左頂部軟組織腫物伴局部骨質缺損,MR顯示左頂部團塊影,與周圍骨質界限不清,呈不均勻明顯強化,鄰近腦組織受壓;D~F.骨纖維異常增殖癥,CT可見枕骨右側板障增厚并含囊性低密度影,MR可見枕骨右側板障內局部膨脹性改變,強化掃描呈大片不規則不強化低信號影,邊緣呈輕度線狀強化
圖2 LCH和骨纖維異常增殖癥的影像學表現
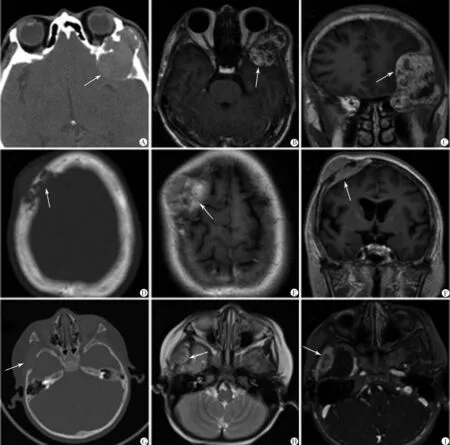
注:A~C.海綿狀血管瘤,CT可見左顳部腫物伴左側額骨、顳骨及眶外側壁膨脹性骨質破壞,MR可見不均勻強化且周邊可見低信號環;D~F.間變性腦膜瘤,CT可見右額骨質破壞,MR可見右額顱骨和頭皮下不規則軟組織腫塊,不均勻強化,鄰近腦膜增厚;G~I.感染性病灶,CT可見右側顳鱗破壞,MR可見右側顱中凹結節狀腫物,界限清晰,近環形顯著強化
圖3 海綿狀血管瘤、腦膜瘤和感染性病灶的影像學表現
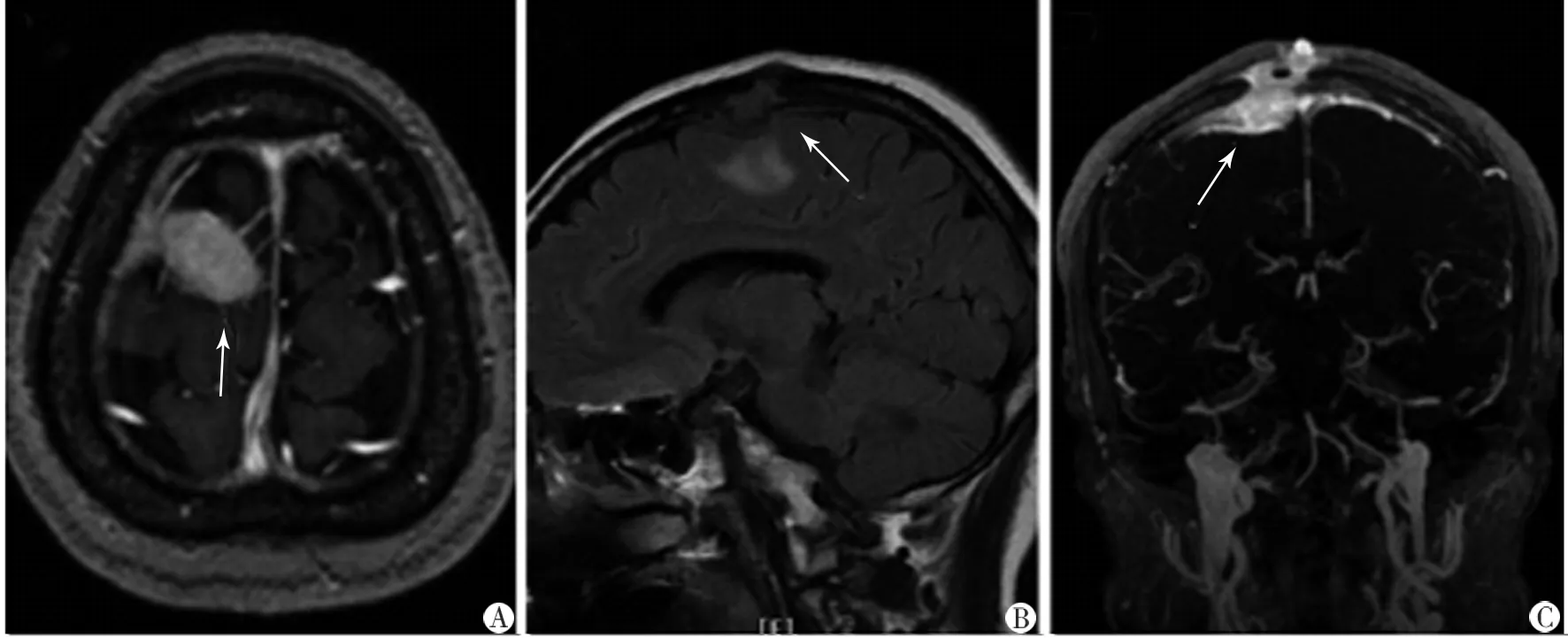
注:A-B.MR可見右頂部中線旁一不規則形結節狀顱內外溝通性腫物,均勻明顯強化;C.MRV可見上矢狀竇受累
圖4 Rosai-Dorfman病的影像學表現
3 討 論
顱蓋骨腫物種類較多,本組34例中就有13種診斷,良性(包括1例感染性疾病)占79.41%(27/34),惡性占20.59%(7/34)。顱蓋骨腫物中,以腫瘤居多,包括原發性腫瘤和繼發性腫瘤,前者起源于顱骨自身,后者包括轉移瘤和起源于鄰近組織繼而侵及顱骨的腫瘤[2-6]。本組資料中,骨瘤是最常見的原發性顱骨腫瘤,占35.29%;轉移瘤是最常見的繼發性顱骨腫瘤,占5.88%。
嗜酸性肉芽腫是LCH的一種表現,后者曾長期被定義為一種全身性非腫瘤性疾病[7],但2013年WHO將LCH定義為中間型(局部侵襲性)骨腫瘤。病因不明,扁骨和長骨常受累,病程較短。顱骨是單發LCH的最常見部位[8],很少侵襲腦組織,影像學上常表現為溶骨性破壞,邊緣清晰,無硬化帶或骨膜反應,可見紐扣樣死骨、地圖樣改變、“雙邊征”;多有軟組織腫塊,可明顯強化,但強化不如腦膜瘤;可伴局部疼痛、血嗜酸性粒細胞增多、低熱、乏力。盡管文獻中有些病例有自限性,但絕大多數需手術治療,甚至需聯合化療和/或放療。有些病例呈難治性,需長期治療和嚴密隨訪,有學者建議應隨訪10年以上。多個器官受累是預后不良的一個危險因素。本組5例中,4例單發,預后良好;1例(典型病例1)呈多灶性且侵襲腦組織,“手術+激素+化療”后仍在第18個月的ECT上發現新病灶。可見,LCH患者的全身ECT、骨髓檢查、綜合治療和長期隨訪必須重視。
R-D病,又名“竇組織細胞增生伴巨淋巴結病(extranodal sinus histiocytosis with massive lymphadenopathy)”,是一種少見的良性特發性非腫瘤性疾病[9]。1969年首次報道,機制尚不清楚,可能與自體免疫系統異常有關。常表現為頸部淋巴結無痛性腫大,只有43%的患者有淋巴結外器官受累。迄今文獻報道中只有100多例累及中樞神經系統[10],多見于矢狀竇旁或顱底,影像學特征不典型,邊界清,伴不規則孔狀骨質破壞,可明顯強化,鄰近腦膜增厚,不易與侵襲硬膜或顱骨的其他腫瘤(如腦膜瘤、嗜酸性肉芽腫、轉移瘤等)鑒別,需組織病理學診斷。有報道認為大多數R-D病有自限性,但其自然史尚不清楚。手術治療仍是合理的選擇,術后可聯合放療或激素治療。本組1例(典型病例2)僅累及中樞神經系統而無淋巴結腫大,罕見,手術切除病灶并輔以激素治療,預后良好。
NHL患者骨骼受累并不少見,但首發于顱骨者少見,約占0.2%[11]。影像學上無特異性表現,難以與其他伴有顱骨或硬膜損害的實體性病變(如腦膜瘤、嗜酸性肉芽腫、轉移瘤等)鑒別,必須取得組織學診斷。由于病例少,尚無標準的治療方案。手術聯合放療、化療仍是目前主要的治療方法。
原發性骨內海綿狀血管瘤(PICH)是一種罕見的、生長緩慢的良性腫瘤,常見于脊柱,但罕見于顱骨,于1845年首次報道,1845年—2015年文獻僅報道93例顱骨PICH[12],多見于顱蓋,尤其額骨,起源于板障內的血管,其機制尚不清楚。臨床表現缺乏特征性,但因含鐵血黃素沉積,在MR的T2加權像上病變周圍常有低信號環,可作為PICH的一個特征性影像學表現。手術是首選的治療方法。
惡性腫瘤顱骨轉移在臨床上較少見,文獻報道中其發生率僅2.3%,容易被誤診或漏診。多見于顱蓋,常多發,表現為穿鑿樣、鼠咬樣溶骨性破壞,邊緣不規則,無硬化帶,鄰近有軟組織腫塊。本組有2例,原發灶或其他器官轉移灶的存在有助于確診。MR在顱骨和腦轉移瘤的篩查上有優勢,ECT有助于查找原發灶和全身轉移灶。
顱蓋骨腫物因其多樣性,且臨床和影像學特征常接近,僅根據臨床和影像學特征有時難以做出正確的診斷,一些少見的或表現不典型的病變會被誤診。本組34例中有11例初步診斷與組織病理學診斷不符,占32.35%,可見,顱蓋骨腫物鑒別診斷的復雜性可能被低估。
合理應用現代影像學技術有助于顱蓋骨腫物的鑒別診斷。良性腫瘤多邊界清楚、有硬化帶、與正常骨質之間的過渡區窄;惡性腫瘤則界限不清、有侵襲性骨膜反應、無硬化帶、過渡區寬泛[2-3]。與X線相比,CT可顯示骨質的細微改變及鈣化,亦可發現部分軟組織病灶。MR能夠清晰顯示軟組織,顯示顱骨受侵襲或破壞程度,對軟組織和腦膜受累程度的顯示優于CT,還可早期發現局限在板障內的病灶,多種序列有助于評估病變性質。
顱蓋骨腫物中,有些具有特征性臨床或影像學表現,如海綿狀血管瘤、LCH、骨纖維結構不良等,較易診斷,但有些因缺乏特征性的臨床或影像學征象而不易鑒別[2-6],如腦膜瘤與NHL或R-D病、惡性纖維組織細胞瘤與腦膜瘤或轉移瘤,需組織病理學檢查方能確診,因此,對于這些鑒別診斷困難的顱骨腫物,建議早期手術治療,明確組織病理學診斷。
總之,多種影像學技術的合理使用,結合患者的年齡、病史和臨床表現,絕大多數顱蓋骨腫物可以正確地鑒別,對于根據臨床表現和影像學難以鑒別或已有骨質破壞者,應及時手術,明確組織病理學診斷。
利益沖突:無
