動物源性食品中β-受體激動劑分類及檢測方法研究進展
2019-10-23 05:45:04廖艷華
食品研究與開發 2019年20期
廖艷華
(廣西壯族自治區疾病預防控制中心,廣西南寧530028)
β-受體激動劑是一類母核為β-苯乙醇胺結構、具有舒張平滑肌及解除支氣管痙攣等作用的藥物,在臨床上被廣泛應用于治療支氣管哮喘。該類藥物能結合細胞膜中的β-受體,從而發生一些列生理效應,減少脂肪沉積,提高瘦肉率[1-2],導致畜禽肉中該類藥物殘留嚴重超標。由于β-興奮劑作用效應較強,并且在動物體內,特別是在動物肝、腎、肺殘留量最高,通過食物鏈進入人體,會使一些易感人群產生較明顯的中毒癥狀,嚴重危害人類健康[3-4]。為保證動物性食品衛生安全,我國農業部已明令[5-6]克倫特羅及其鹽、沙丁胺醇及其鹽、西馬特羅及其鹽等β-受體激動劑為禁止使用藥物,不得在所有動物性食品中檢出,并制定了相應的監管制度與檢測方法[7-12]。
1 β-受體激動劑種類
β-受體激動劑大多含苯乙醇胺骨架,按苯環取代基不同可分為與胺基、酚羥基、鹵素等各種取代基鏈接;根據胺基上的取代基不同,可分為叔丁基或異丙基類興奮劑,這有利于β-受體激動劑裂解途徑及機理分析、其二級質譜豐富的特征離子碎片信息更有利于目標物的篩查、確證。在國內最常用的有克倫特羅、沙丁胺醇、特布他林等。
β-受體激動劑化學結構見圖1。
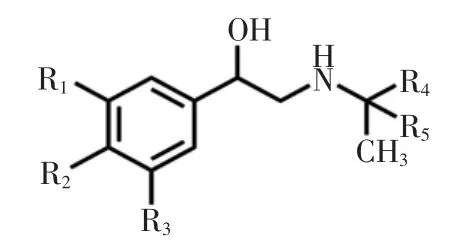
圖1 β-受體激動劑化學結構Fig.1 Molecular structural of β-agonist
β-受體激動劑常見種類及特征基團見表1。
2 β-受體激動劑的檢驗方法
β-受體激動劑的檢測方法包括依據其不同樣品基質前處理方法和檢測儀器的不同,目前已建立了液相色譜、酶聯免疫法、氣相色譜串聯質譜法(gas chromatography-mass spectrometer,GC/MS)、液相色譜串聯質譜法(high performance liquid chromatography-mass spectrometer HPLC-MS/MS),由單組分檢測逐漸向多組分同時檢測發展,靈敏度也逐步提高。
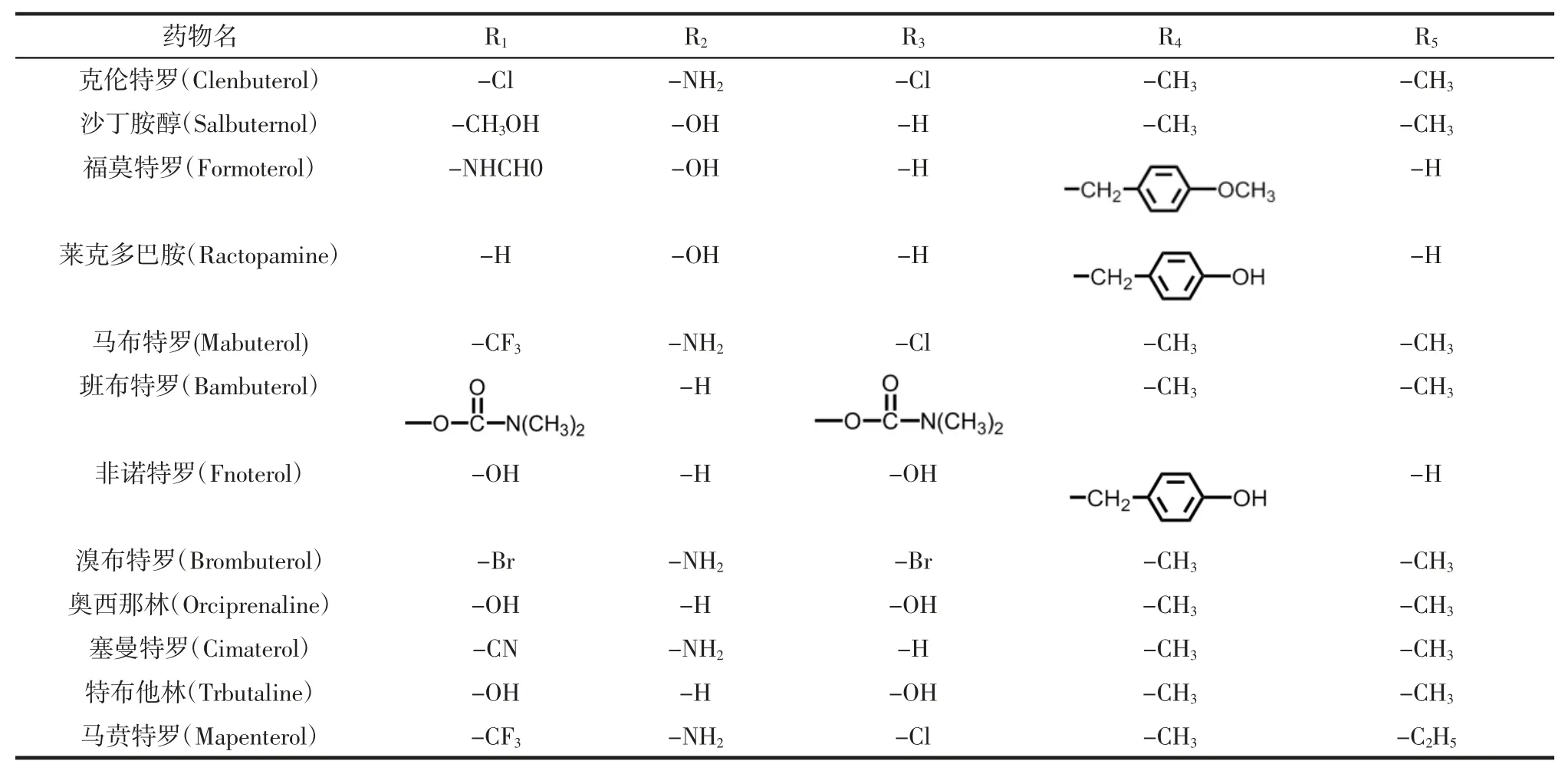
表1 β-受體激動劑常見種類及特征基團Table 1 Common species and characteristic groupsof β-agonist
2.1 高效液相色譜法
目前現行有效的國家標準方法GB/T5009.192-2003《動物性食品中克倫特羅殘留量的測定》[12]第一法為液相色譜法,此法僅為單個組分克倫特羅的檢測,試樣剪碎后用高氯酸勻漿,進行超聲加熱提取后,用異丙醇 ∶乙酸乙酯=40 ∶60(v/v)萃取,有機相濃縮,經弱陽離子交換柱進行分離,用乙醇∶氨=98 ∶2(v/v)溶液洗脫,洗脫液經流動相定容后在高效液相色譜儀上紫外檢測器進行測定,外標法定量,檢出限為0.5 ug/kg。王爍等[13]建立了超高效液相色譜測定豬組織中4 種β-興奮劑(奧西那林、克倫特羅、沙丁胺醇、萊克多巴胺)的方法,流動相采用乙腈和0.01 mol/L 磷酸二氫鉀溶液的梯度洗脫,檢測波長為227 nm,外標法定量,4 種興奮劑的檢出限為 0.1 μg/kg~0.4 μg/kg,不同加標水平回收率為61.2%~101.3%,相對標準偏差分別為2.9%~9.2%,此法在分離能力、分析速度及靈敏度方面都比高效液相色譜法(high performance liquid chromatography,HPLC)有較大提高,二極管陣列檢測器(photo-diode array,PDA)可以提供 190 nm~400 nm 波長的吸收曲線圖,在保留時間定性基礎上再結合標準物質與樣品中待測物吸收曲線匹配度定性。但高效液相法依然存在靈敏度低、無法同時進行多種興奮劑的測定等缺點,而且存在較多雜質干擾,給定性確證帶來困難,因此在近些年應用得很少。
2.2 免疫分析法
免疫分析法是以抗原與抗體的特異性、可逆性結合為基礎檢測各種物質的分析方法,由于免疫法選擇性較好,具有前處理簡單、靈敏度高等優點,故在快速篩查大批量樣品中獸藥殘留方面有廣泛應用,免疫分析法中最常用的是酶免疫分析法和膠體金免疫層析法。
酶免疫分析法其原理是待測樣品中的β-受體激動劑類藥物殘留與微孔包被的抗原共同競爭特異性抗體。萬宇平等[14]應用直接競爭酶聯免疫吸附技術,建立了一種快速檢測動物組織中β-興奮劑類藥物多殘留的方法,并對其技術性能進行了評價。結果表明,該方法的IC50動范圍為0.4 μg/L~0.7 μg/L,對組織樣本的檢測限為0.5 μg/kg;樣本添加回收率為72.9%~103.5%,變異系數為5.8%~14.1%;可用于檢測克侖特羅、沙丁胺醇等8 種β-受體激動劑。張璐琪等[15]用比較了3 種不同公司生產的試劑盒測定3 種β-興奮劑殘留,結果顯示鹽酸克倫特羅試劑盒和萊克多巴胺試劑盒的檢出結果與液質法的符合率較高,均為94%;沙丁胺醇試劑盒和β-興奮劑試劑盒的符合率一般,分別為76.2%和75%,主要表現在假陽性結果較多。龔倩等[16]建立了快速檢測豬肉中克倫特羅、沙丁胺醇、溴布特羅和特布他林4 種β-受體激動劑的光激化學發光納米均相時間分辨熒光免疫(alpha LISA)分析方法,沙丁胺醇與克倫特羅、溴布特羅和特布他林的交叉反應率分別為143.3%、179.2%和95.6%,檢出限為0.03 ng/mL~0.08 ng/mL,在 0.5、10、20 μg/kg 3 個添加水平下,4 種化合物的平均回收率為82.5%~15.9%,相對標準偏差均小于12.0%。
膠體金免疫層析法是一種利用膠體金顆粒作為標記物簡單快速的免疫分析方法[17]。其最關鍵的是制備出特異性強和純度高的單克隆抗體,使用原理是將人工合成的抗原先固定于條狀纖維層析材料的試紙條上,膠體金標記試劑(抗體或單克隆抗體)吸附在結合墊上,樣品溶液借助毛細作用在試紙條上移動,待測物與人工合成的抗原競爭結合膠體金標記的抗體,并直接以顏色顯示檢測結果。目前主要用在液體樣品檢測中,在肉制品中應用不多,其靈敏度和準確度不及酶聯免疫法。
現行有效標準免疫方分析法[18-19]主要是飼料和動物尿液中受體激動劑的酶聯免疫試劑盒方法。GB/T 5009.192-2003《動物性食品中克倫特羅殘留量的測定》第三法為酶聯免疫法。
雖然免疫法操作簡單,使用成本不高,在監測檢測中能快速測量大批量樣品,但免疫學存在交叉反應現象仍然是制約其應用的主要問題,而且受廠家生產試劑質量影響,有可能抗體批次不同,測定結果也會出現差異。另外在同類藥物多殘留檢測方法方面也有其不能所及之處,不能用于國家相關執法仲裁檢驗。
2.3 色譜質譜聯用技術
色譜聯用技術即兩種或兩種以上的分析技術聯合應用,以達到更好的分析效果。色譜質譜聯用技術一方面具有色譜的分離能力、分析速度和靈敏度,又兼具質譜對化合物的結構的定性能力,因而在檢測的選擇性和準確度上都有更好表現。色質聯用技術是目前分析β-受體激動劑殘留的主流方法,包括氣相色譜-質譜聯用(gas chromatography-mass spectrometer,GC-MS)和液相色譜-質譜聯用(HPLC-MS)。我國現行檢驗方式依據不同樣品基質前處理方法及檢測儀器制定,對 GC-MS[10,12]和 HPLC-MS[7,8,9,11]檢測方法做了相關規定和闡述。
受體激動劑的GC/MS 分析與其他分析方法不同主要是樣品需衍生化,因為β-受體激動劑屬于高沸點、難揮發的極性化合物,不適合直接進行GC 或GCMS 分析。通過衍生化,不僅可以增大試樣的揮發性和穩定性,減小樣品的極性,還可以達到改善分析效果、提高靈敏度的目的。衍生反應的類型目前文獻報道的主要有3 類:第一類為酰化反應,衍生劑通常為七氟乙酰酐、乙酸酐;第二類為甲基硼酸或丁基硼酸化反應,利用β-受體激動劑結構的側鏈上有-OH 和-NH-與衍生試劑形成五元環,由于硼的同位素為很強,故其衍生物的質譜峰很有特征性。第三類采用硅烷化反應,衍生劑通常為雙三甲基硅基三氟乙酰胺(bis(trimethylsilyl)trifluoroacetamide,BSTFA)+1 %(φ)三甲基氯硅烷(trimethyl chlorosilane,TMCS)衍生,最后一種是最常用的衍生劑。報道的相關文獻[20-23]和標準方法[10,12]GC-MS 樣品前處理大多都是樣品經過酶解或酸解之后,用pH5.2 的乙酸鈉緩沖溶液或酸性甲醇或高氯酸溶液提取樣品中的獸藥殘留,經高氯酸沉淀蛋白后,堿性條件下經過乙酸乙酯 ∶異丙醇(60 ∶40,v/v)進一步提取凈化、濃縮,在pH5.2 緩沖體系下經固相萃取柱凈化,最后衍生經GC-MS 內標法定量測定;劉五一等[24]用氣質法測定畜禽肉中4 種β-興奮劑的前處理新技術進行研究,結果表明直接用5%的高氯酸提取,WCX 陽離子交換固相萃取小柱效果最佳,檢測的4 種興奮劑在 50 μg/L~1 000 μg/L 的范圍具有良好線性,加標回收率在84.32 %~103.15 %之間,RSD 在1.35%~4.78%之間。岳韓笑等[25]采用同位素稀釋質譜法,結合HLB-MCX 雙柱固相萃取技術,建立了豬肉中克倫特羅、妥布特羅、溴布特羅、沙丁胺醇等4 種β-受體激動劑殘留量的氣相色譜-串聯質譜(GC-MS/MS)檢測方法,方法最低檢測限為0.13 μg/kg~0.40 μg/kg,最低定量限為0.40 μg/kg~1.27 μg/kg。由于這種方法衍生耗時長,條件不好控制,同時能檢測的受體激動劑不是很多,因此方法的使用在逐年減少。
相比于GC-MS、HPLC-MS/MS 的前處理相對簡單,不需要經過衍生化步驟,因此國內得到快速普及,現行主流的適用于動物源性β-食品受體激動劑的LC/MS/MS 國家標準分析方法只要有3 個,分別是GB/T 21313-2007 《動物源性食品中β-受體激動劑殘留檢測方法液相色譜-質譜/質譜法》、農業部1025 號公告-18-2008《動物源性食品中β-受體激動劑殘留檢測液相色譜-串聯質譜法》、GB/T 22286-2008《動物源性食品中多種β-受體激動劑殘留量的測定液相色譜串聯質譜法》。
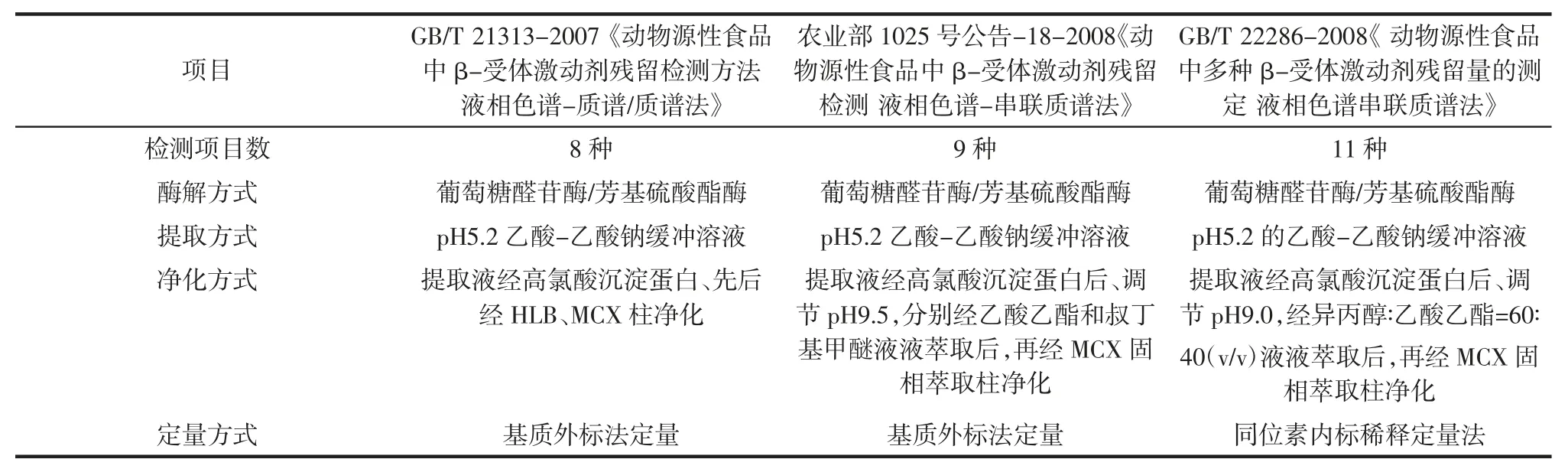
表2 3 種現行β-受體激動劑國家標準檢驗方法比較Table 2 Comparison of national standard methods of 3 β-agonist
表2 列舉了3 種國家標準檢驗方法的檢驗步驟及定量方式對比發現,3 種方法的標準分析方法各有優缺點,GB/T 21313-2007《動物源性食品中β-受體激動劑殘留檢測方法液相色譜-質譜/質譜法》的檢驗方法能測定8 種β-受體激動劑,提取液不用調節pH 值可直接凈化,但是本法需經雙柱凈化,兩次溶劑揮干,檢測成本高,過程耗時;基質外標法定量檢測結果受檢驗人員操作影響較大;不同類型的食品其基質效應也不一樣,針對分析樣品的基質還需配制不同基質標準曲線,增大了檢驗工作量;農業部1025 號公告-18-2008 檢驗方法能測定9 種β-受體激動劑中,用一步液液萃取代替固相萃取凈化,雖然節省檢測成本,但需調節pH 值,人員勞動強度增大,大量溶劑的揮干容易對環境產生污染,同樣也存在基質外標法定量無法準確等缺點;GB/T 22286-2008《動物源性食品中多種β-受體激動劑殘留量的測定液相色譜串聯質譜法》檢驗方法能測定11 種β-受體激動劑,采用基質內標稀釋法定量模式,能減少因為樣品提取、質譜進樣和離子化等多因素造成的差異,定量結果更準確,但液液提取時用的提取溶劑為異丙醇∶乙酸乙酯=60 ∶40(v/v),由于異丙醇在提取液中的比例高、沸點高,其在揮干中需要耗費大量時間,如完全按國標方法操作,異丙醇 ∶乙酸乙酯=60 ∶40(v/v)揮干至少需要 4 h,這給檢測過程造成了很大的困擾。
近些年對于液質聯用法檢測β-受體激動劑,大多學者研究的方向集中在樣品的前處理技術與儀器多殘留檢測技術的開發應用上,能同時分析11 種~35 種受體激動劑。前處理研究的重點主要集中在樣品水解方式的選擇及凈化方法的優化上。賈玉珠等[26]和蔡增軒[27]建立了超高效液相色譜一電噴霧串聯四極桿質譜法檢測動物組織中20 種β-興奮劑殘留,20 種β-興奮劑殘留用3 種同位數內標進行定量。受體激動劑在55 ℃經葡萄糖苷酸酶酶解2 h,此酶解方式較傳統或國標37 ℃酶解16 h 節省了很多時間。劉先軍等[28]用1 %三氯乙酸溶液超聲提取15 min 動物組織中26 種β-受體激動劑,認為此酸水解的方式起到了水解軛合物的作用,同時具有提取待測物、沉淀蛋白蛋白功能,相對酶解方比較,減少了酶水解中酶制劑本身及其酶解產物對目標物的干擾,滿足快速檢測化的要求。凈化方式優化研究方向主要集中在固相萃取技術上,張瑞雨等[29]優化了樣品前處理方法,應用于高效液相色譜-串聯質譜(HPLC-MS/MS)法檢測豬肉、豬肝中萊克多巴胺、沙丁胺醇β-受體激動劑。沙丁胺醇和萊克多巴胺的檢出限分別為0.13、0.14 μg/kg,在不同基質濃度下的相對標準偏差分別為5.9%~16.2%和6.4%~20.2%。該方法與GB/T 22286-2008《動物源性食品中多種β-受體激動劑殘留量的測定液相色譜串聯質譜法》的預處理方法比較,使用鹽酸代替高氯酸調節pH2.0,達到沉淀蛋白與樣品過柱凈化前調節pH 值雙重效果,凈化樣品時減少了有機溶劑的使用,用價格稍低的SCX 凈化柱代替MCX 凈化柱,洗脫液用5%氨化乙酸乙酯代替5%氨化甲醇,節約了成本,提高了檢驗效率,減少了工作量。馬俊美等[30]建立在線固相萃取-高效液相色譜-串聯質譜法測定豬肉和羊肉中10 種β-受體激動劑的全自動分析方法,樣品經MCX在線固相萃取小柱凈化,XBridge C18色譜柱分離,檢出限為 0.004 μg/kg~0.040 μg/kg;方法回收率為 76.5%~107.7%,在線固相萃取提高了樣品的自動化程序,保證了方法重現性,提高工作效率。
除了傳統的液液萃取與離子交換固相萃取的凈化模式,張學亮等[31]建立了一種分子印跡固相萃取-超高效液相色譜-串聯質譜同時測定豬肉中5 種β-受體激動劑殘留的方法,檢出限為 0.005 μg/kg~0.009 μg/kg,添加水平為 0.25、1、5 μg/kg 時,回收率為 80.4%~92.9%,相對標準偏差為1.2%~6.3%。史娜等[32]建立MPI 固相萃取-同位素稀釋法-高效液相色譜串聯質譜測定動物源性食品中35 種β-受體激動劑和11 種β-受體阻斷劑殘留的方法,前處理方法大多與GB/T 22286-2008《動物源性食品中多種β-受體激動劑殘留量的測定液相色譜串聯質譜法》雷同,不同之處是液液萃取的異丙醇 ∶乙酸乙酯比例為 1 ∶1(v/v),用 MPI 固相萃取代替MCX 固相萃取,由于檢測數目較多,用的內標多達10 種,保證了結果的準確性。羅輝泰等[33]建立了分散固相萃取-同位素稀釋-高效液相色譜-串聯質譜(dSPE-ID-HPLC-MS/MS)同時測定豬肉中26 種β-受體激動劑殘留的方法,樣品經酶解、乙酸銨緩沖液提取、高氯酸沉淀蛋白、離心后,調節pH9.0,乙腈反萃取后經凈化上機測定。檢出限分別為0.03 μg/kg~0.1 μg/kg,在3 個不同濃度的添加水平下,平均回收率為65.3%~108.5%,相對標準偏差為2.7%~13.3%,通過同位素內標進行校正后其回收率能滿足殘留分析檢測要求,該方法成本低,不用經過復雜的固相萃取步驟,大大縮短了樣品前處理時間。李磊等[34]建立QuEChERS EMR-Lipid 為前處理技術測定動物源性食品中8 種β-受體激動劑,樣品經酶解、離心處理后,先后經過增強型脂質快速凈化管(QuEChERS EMR-Lipid)、脂質凈化反萃取管(QuEChERS Final Polish EMRLipid)凈化后上LC-MS/MS 分析測定,8 種組分的檢出限范圍為0.2 μg/kg~0.4 μg/kg,加標回收率為 79.1%~94.8%。
近年來隨著檢測儀器的發展,基于高分辨質譜可以測定化合物的精確相對分子量,對樣品進行高靈敏度、高質量精度的全掃描分析,對復雜樣品中目標化合物的檢測具有明顯優勢。因此成為近年動物源性食品中高通量獸藥殘留篩查與確證研究的熱點,劉暢等[35]建立豬肉中31 種β-受體激動劑的高效液相色譜-四級桿-飛行時間質譜(HPLC-Q-TOF-MS)檢測方法,31 種化合物的檢出限為 0.01 μg/kg~5 μg/kg,結合自建的精確相對分子質量數據庫,通過比較精確相對分子質量、保留時間、同位素峰以及特征碎片離子等信息,進行篩選檢測,適合于動物源性食品中可能存在的β-受體激動劑的篩查。
2.4 其他檢測方法
激動劑其他檢測方法還有毛細管電泳法[36]、生物傳感器法,但這些方法目前不是特別的完善法,實際工作中很少用到,存在各自的缺點,需要進一步研究。
3 結論
通過研究發現,HPLC-MS/MS 依然是動物源性食品中定性定量分析檢測的主要手段,但由于不同食品基質的復雜,前處理步驟多、耗時長以及分析存在較強基質效應的影響,其方法靈敏度、精密度及基質效應的大小受技術人員操作影響較大,因此,對于β-激動劑的殘留檢測,仍需從4 個方面著手:1、加快現行國家標準的整合、補充和完善。由于現行的多個國家標準檢驗方法存在著部分交叉、重合,涉及的檢測項目種類為19 種,也沒有完全涵蓋國家相關法規或制度所羅列的全部項目,無法保證相關法規的有效實施;2、為了滿足食品安全風險監測檢測技術高通量、快速化分析技術要求,待測物提取和凈化步驟的改進依然是未來今年研究熱點,因此認為未來更有效、更簡化的凈化模式是前處理研究的重點,其中最能實現該目標的前處理方法認為是QUEChERS 和SPE 技術,這兩種技術的優勢都包括低試劑消耗及高通量,雖然QUECh-ERS 技術主要針對提取液中的雜質,凈化效果不及傳統SPE,但筆者認為它在低試劑消耗、高通量的多殘留篩查方面仍有明顯優勢。3、三重四極桿質譜有很高的靈敏度,很好的線性范圍,對于監控目標物是非常有效的利器。但是近些年,發生食品安全問題的往往是那些不在監控清單中的物質,食品安全未知物監控領域將是未來的技術發展方向,因此更有效的質譜掃描模式及其分析軟件、飛行時間和軌道阱等高分辨質譜對低殘留樣品的定性定量、多殘留篩查以及物質精確質量數的測定和化合物結構研究方向方面提供更為有力的檢測工具。4、由于精密儀器價格昂貴、試劑耗材用量大、檢測過程復雜、技術要求高、檢測時間長等因素,因此建立特異性強、靈敏度高免疫分析快速篩查方法可有效提高檢驗效率,在現場監控和大規模篩選檢測中仍有廣闊的應用前景。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
