固相萃取LC-MS/MS法測定疏血通注射液中次黃嘌呤濃度*
2019-10-24 06:29:58王獻瑞趙西子王曉明郭亞卿潘桂湘黃宇虹
天津中醫藥大學學報 2019年5期
王獻瑞 ,趙西子 ,王曉明 ,郭亞卿 ,潘桂湘 ,黃宇虹
(1.天津中醫藥大學,天津市現代中藥重點實驗室-省部共建國家重點實驗室(培育),天津 301617;2.天津中醫藥大學第二附屬醫院,天津 300250)
疏血通注射液是由水蛭[1-2]、地龍[3-4]兩味動物類中藥經低溫提取和膜分離等工藝制成的中藥復方制劑[5],具有活血化瘀、通經活絡的功效,臨床常用于瘀血阻絡所致的缺血性中風病中經絡急性期,癥見半身不遂、口舌歪斜、語言謇澀[6-7]。疏血通注射液化學成分復雜,含有多肽、多糖、氨基酸、次黃嘌呤等物質[8-10],目前其真正發揮藥效作用的物質基礎尚不明確。據文獻報道[11-12],次黃嘌呤具有明確的生物活性,能透過細胞膜進入細胞內,提高多種酶的活性,參與機體一些重要生理機能的調節。因此,本文以次黃嘌呤為指標成分,建立液相色譜串聯質譜(LC-MS/MS)法對其進行定量分析,以期為疏血通注射液質量分析、評價以及質量標準的制定提供參考和依據。
目前,疏血通注射液中次黃嘌呤的測定,采用的大多是高效液相色譜-紫外法(HPLC-UV)[13-14],檢測波長為254 nm。由于疏血通注射液中眾多成分在254 nm處具有紫外吸收,需進行長達50 min的梯度洗脫,使次黃嘌呤與注射液中的其他干擾物質實現完全的色譜分離,次黃嘌呤出峰時間為13min,整個分析周期相對較長[15]。LC-MS/MS具有高選擇性、高靈敏度、高準確性的特點[21],尤其是具有獨特的質量分辨能力,能在復雜基質中專屬、靈敏、準確地檢測某個或某幾個成分。本研究采用固相萃取LC-MS/MS法測定疏血通注射液中次黃嘌呤濃度,分析時間縮短至3.50 min,次黃嘌呤出峰時間為1.02 min,可較大程度提高分析的效率。
1 儀器與試藥
1.1 儀器 Waters ACQUITY UPLC超高效液相色譜儀(美國waters公司),Waters Xevo TQ-S三重串聯四極桿質譜儀(美國waters公司);MiniSpin Plus高速離心機(德國Eppendorf公司),AX205十萬分之一電子天平(瑞士Mettler Toledo公司),Synergy UV超純水機(美國Millipore公司)。
1.2 試劑和藥品 疏血通注射液(牡丹江友博藥業股份有限公司)。次黃嘌呤標準品(德國Sigma公司,純度99%以上),6-巰基嘌呤標準品(德國Aldrich公司,純度98%以上)。甲醇(色譜純,德國Sigma公司),甲酸(色譜純,ROE公司),超純水為Millipore超純水系統制得。Sep-pak C18固相小柱(500 mg,waters公司)。
2 方法與結果
2.1 色譜條件 Waters ACQUITY UPLC超高效液相色譜儀系統:二元泵系統、自動進樣器、柱溫箱,Acquity UPLC BEH C18(100 mm×2.1 mm,1.7 μm)分析柱,BEH C18(5 mm×2.1 mm,1.7 μm)保護柱;流動相為甲醇(A),0.1%甲酸水(B),流速:0.3 mL/min;柱溫40℃,進樣量2 μL,分析時間為3.50 min。梯度洗脫程序見表1。
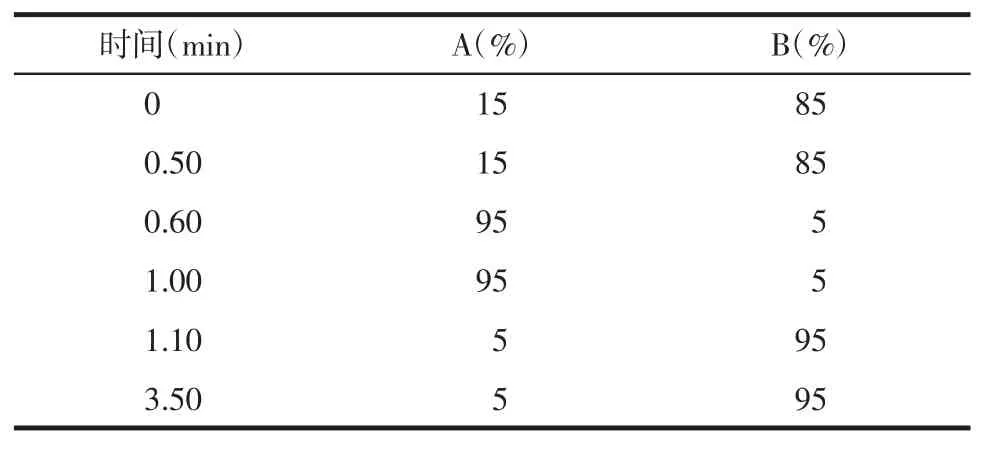
表1 色譜條件梯度洗脫程序表
2.2 質譜條件 Waters Xevo TQ-S三重四極桿質譜,ESI負離子模式,MRM掃描方式。參數設置:毛細管電壓3.0 kV,錐孔電壓30 V,氮氣壓力7 psi,脫溶劑氣溫度400℃,脫溶劑氣流速800 L/h,錐孔氣流速150 L/h,碰撞氣流速0.15 mL/min。化合物MRM方法參數見表2。
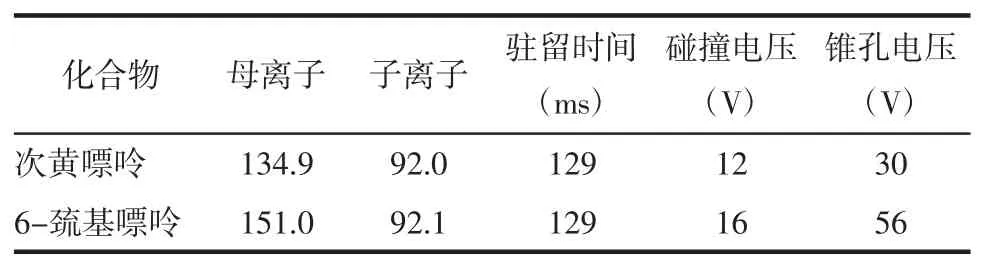
表2 次黃嘌呤與內標的MRM方法參數
2.3 溶液的制備
2.3.1 對照品溶液制備 精密稱取次黃嘌呤適量,加甲醇制成100 μg/mL的次黃嘌呤儲備液;另稱取內標6-巰基嘌呤適量,少量甲醇溶解后用甲醇-水(含0.2%甲酸)(2∶8) 配制成 125 μg/mL 的 6-巰基嘌呤儲備液。用甲醇-水(2∶8,含0.2%甲酸)稀釋上述儲備液,制得含次黃嘌呤系列濃度 500、400、300、200、100 ng/mL的混合對照品溶液(含內標250 ng/mL)。2.3.2 供試品溶液制備 精密移取100 μL疏血通注射液和100 μL濃度為125 μg/mL的6-巰基嘌呤溶液,加于已活化的C18固相萃取小柱上,用300 μL水洗除雜,再用 1.00 mL 甲醇-水(2∶8,含 0.2%甲酸)洗脫,取洗脫液 100 μL 用甲醇-水(2∶8,含 0.2%甲酸)稀釋 10倍,搖勻,再取 200 μL用甲醇-水(2∶8,含0.2%甲酸)稀釋5倍,混勻,即得。
2.3.3 陰性對照液制備 取1 mL疏血通注射液,加黃嘌呤氧化酶37℃水浴溫孵2h,精密移取100μL經酶促反應后的疏血通注射液,加于C18固相萃取小柱上,其余按“2.3.2”項下方法操作,制成不含次黃嘌呤的陰性對照液。
2.4 專屬性試驗 取陰性對照液、對照品溶液和供試品溶液分別進樣測定,次黃嘌呤、6-巰基嘌呤色譜峰形良好,測定無干擾,次黃嘌呤保留時間為1.02 min,6-巰基嘌呤保留時間為1.22 min,結果見圖1。
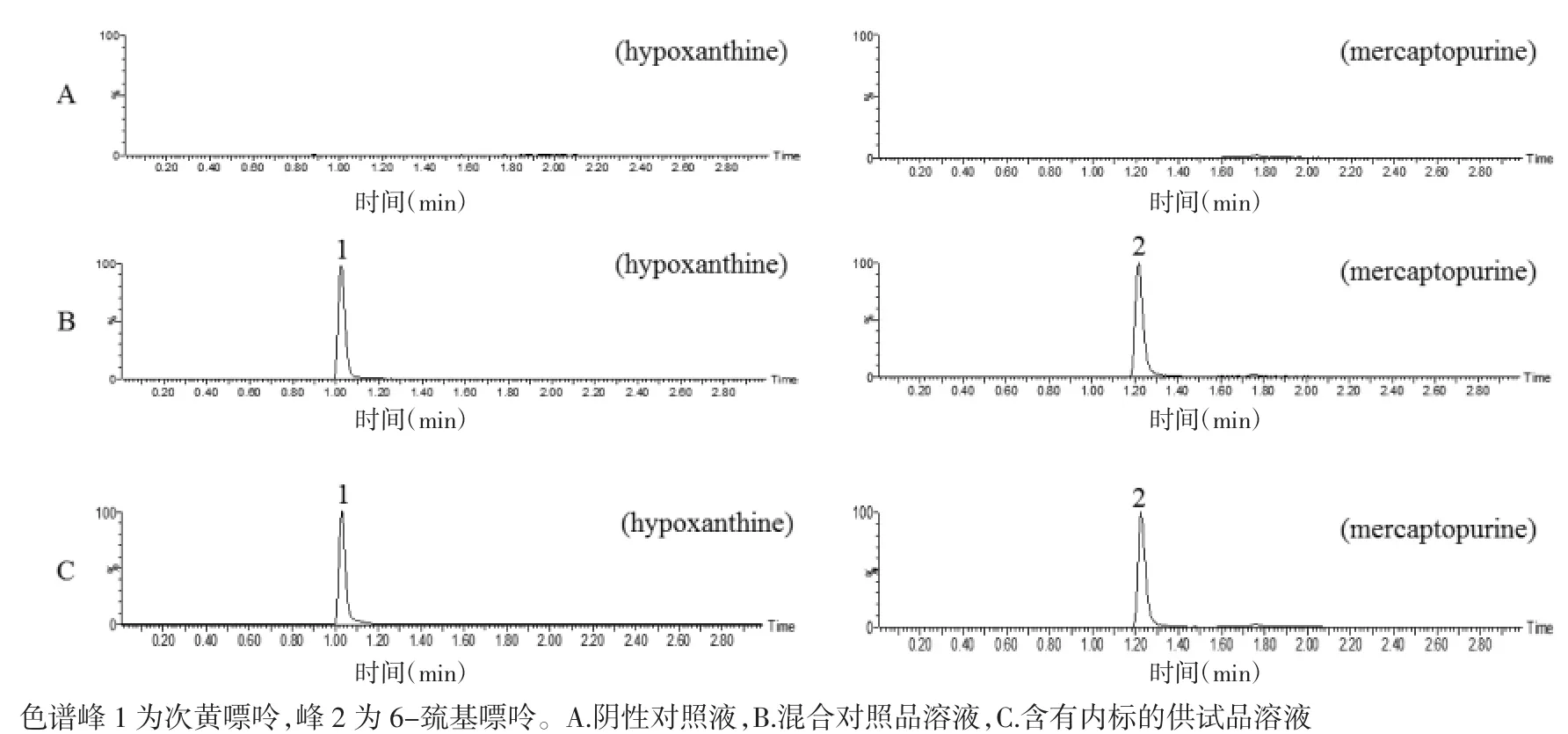
圖1 疏血通注射液中次黃嘌呤及內標的MRM色譜圖
2.5 線性關系考察 吸取“2.3.1”項下系列濃度混合對照品溶液,進樣2 μL測定,以次黃嘌呤質量濃度(X)為橫坐標,待測物與內標的峰面積比值(Y)為縱坐標繪制標準曲線,進行線性回歸,得回歸方程Y=4.299×10-3X+9.246×10-3,r=0.999,表明次黃嘌呤在0.1~0.5 μg/mL范圍內線性關系良好。
2.6 精密度實驗 精密吸取對照品混合溶液2 μL,按“2.1”項下色譜條件重復進樣6次,測得次黃嘌呤與內標峰面積比值,相對標準偏差(RSD)為0.54%,表明儀器精密度良好。
2.7 重復性實驗 精密移取同一批號的疏血通注射液 100 μL,按“2.3.2”項制備供試品溶液,平行6份,進樣測定,RSD為0.86%,表明樣品重現性良好。
2.8 穩定性實驗 室溫下,精密吸取同一供試品溶液 2 μL,分別在 0、2、4、6、8、12、24、48 h 進樣,測次黃嘌呤與內標峰面積比值,RSD為0.84%。結果表明供試品溶液在48 h內穩定。
2.9 加樣回收率實驗 精密移取同一批號已知含量的疏血通注射液6份,每份50 μL,精密加入濃度為 200 ng/mL 的對照品溶液 28 μL,按“2.3.2”項下處理與測定,計算回收率,結果見表3。
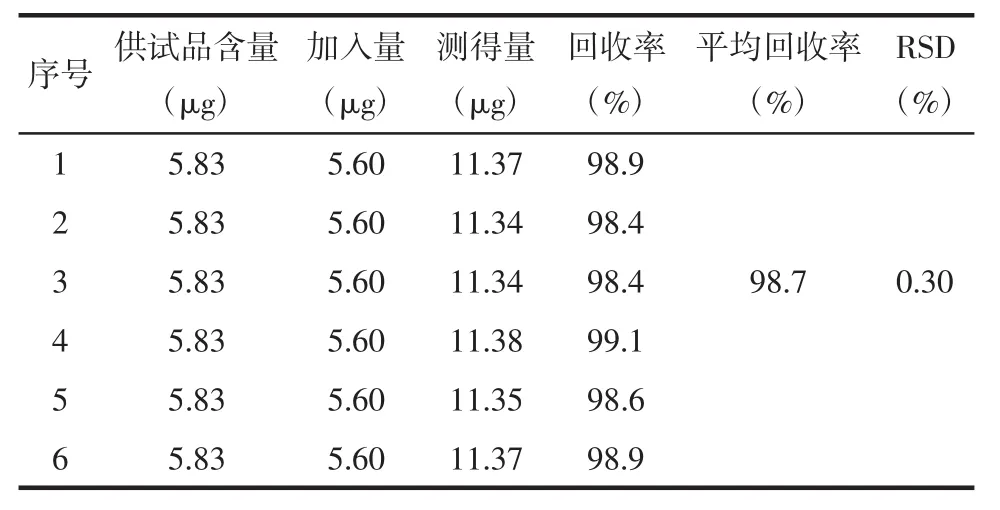
表3 次黃嘌呤加樣回收率試驗結果(n=6)
2.10 樣品濃度測定 分別精密移取各批號疏血通注射液 100 μL,按“2.3.2”項下方法制備供試品溶液,內標法計算次黃嘌呤濃度,結果見表4。
3 討論
3.1 流動相的選擇 實驗分別考察了乙腈-水和甲醇-水等流動相系統,結果發現當使用乙腈-水或乙腈-水(含0.1%甲酸)為流動相時,次黃嘌呤峰形尖而細,但主峰后出現一個小的裂分峰;當使用甲醇-水為流動相時,次黃嘌呤峰形較寬,且裂分為兩個大小相當的雙峰,但若將水相改為0.1%甲酸水時,即以甲醇-水(含0.1%甲酸)為流動相,峰形對稱尖銳且未出現裂分,響應較高。當以甲醇-水(含0.2%甲酸)為流動相時,其峰形和響應與甲醇-水(含0.1%甲酸)相當,考慮到色譜柱對酸的耐受程度,實驗最終選擇甲醇-水(含0.1%甲酸)作為流動相。

表4 疏血通注射液中次黃嘌呤濃度測定結果
3.2 稀釋溶劑的選擇 在供試品和對照品溶液制備過程中,發現終溶液如果所含甲醇比例較高,會因與流動相不匹配而產生強溶劑效應,導致色譜峰形的畸變;反之,如果采用純水稀釋系列溶液,則會因為內標在水中的溶解度低而析出形成渾濁液體。實驗考察比較了甲醇-水系統(水相不含,或含有0.1%甲酸、0.2%甲酸、0.3%甲酸) 不同比例 5∶5、4∶6、3∶7 和2∶8作為稀釋劑時,次黃嘌呤和內標6-巰基嘌呤的峰形和響應,結果表明當以甲醇-水(2∶8,含0.2%甲酸)為稀釋劑時,二者的響應最高且峰形良好。
3.3 離子源的選擇 考察了待測物在ESI和APCI兩種電離方式下的響應,發現采用APCI電離源,次黃嘌呤和6-巰基嘌呤的響應極其微弱,而采用ESI電離源二者的響應提高了幾個數量級,這可能與次黃嘌呤和6-巰基嘌呤均為極性化合物有關,更適合選用ESI電離源。
3.4 電離方式的選擇 將次黃嘌呤和6-巰基嘌呤對照品溶液經由蠕動泵連續進樣,比較正、負電離方式下化合物的響應,發現次黃嘌呤和6-巰基嘌呤在負離子模式下,均可產生響應較高且穩定的[MH]-離子。因此選擇在負離子模式下采用MRM掃描方式檢測。
3.5 實驗結果分析 本文建立的分析方法可方便、準確測定疏血通注射液中次黃嘌呤濃度,為其提供技術支持,較常規UV檢測方法可大大縮短分析時間。國家食品藥品監督管理局頒布的WS3-548(Z-084)-2005(Z)文件,要求疏血通注射液中含次黃嘌呤不得低于0.05 mg/mL,由表4可見,20個批次疏血通注射液中次黃嘌呤濃度穩定集中在0.117~0.138 mg/mL,即(0.129±0.005)mg/mL(n=20),表明產品質量合格、生產工藝穩定,次黃嘌呤可作為疏血通注射液質量分析、評價的指標成分。
4 結論
本文所建立的固相萃取LC-MS/MS方法簡便、準確、靈敏,可用于疏血通注射液中次黃嘌呤濃度測定。
