UPLC指紋圖譜結合化學模式識別評價白芷藥材質量
2019-11-05 09:15:10唐志書劉妍如宋忠興孔馨逸魏思敏
天然產物研究與開發 2019年10期
陳 琳,唐志書,劉妍如,宋忠興,孔馨逸,魏思敏,孫 琛,鞠 浩
1陜西省中藥資源產業化協同創新中心 秦藥特色資源研究開發國家重點實驗室(培育) 陜西省創新藥物研究中心,咸陽 712083;2陜西中醫藥大學藥學院,西安 712046
白芷為傘形科植物白芷Angelicadahurica(Fisch.ex Hoffm.)Benth.et Hook.F或杭白芷A.dahurica(Fisch.ex Hoffm.)Benth.et Hook.F.var.formosana (Boiss.)Shan et Yuan的干燥根,其性辛、溫,歸胃、大腸、肺經,始載于《神農本草經》[1]。臨床應用廣泛,具有解表散寒、祛風止痛、宣通鼻竅、燥濕止帶、消腫排膿之功效,用于治療感冒頭痛、眉棱骨痛、鼻塞流涕、帶下、瘡癰腫痛等癥[2]。
呋喃香豆素是白芷中的主要活性成分,《中國藥典》2015年版規定白芷干燥品中含歐前胡素的含量不得少于0.08%[2]。除歐前胡素外,白芷中還含有水合氧化前胡素、異歐前胡素、白當歸素、氧化前胡素、佛手柑內酯等呋喃香豆素成分[3,4]。現代藥理學研究表明,白芷及其活性成分具有抗炎、鎮痛、抗氧化、抗腫瘤以及抗心腦血管疾病等多種藥理活性[5-9]。
四川、杭州、安徽、河南以及河北等地是白芷的主要產地,分別為川白芷、杭白芷、亳白芷、禹白芷和祁白芷,不同產地白芷的化學成分有較大差異[10]。除產地之外,加工方法是影響白芷藥材質量的另一個重要因素,據報道,經硫磺熏蒸后,呋喃香豆素類成分的含量顯著下降[11],鎮痛活性也顯著降低,表明白芷活性成分易受產地、加工方式等的影響。目前對白芷質量控制的研究,主要集中在歐前胡素、異歐前胡素等幾個呋喃香豆素成分的含量測定,也有白芷藥材HPLC指紋圖譜的研究[12,13],但均未能較全面表征白芷藥材成分及影響各批次白芷藥材差異的標志性成分。
指紋圖譜和化學識別模式是評價中藥質量的重要方法,指紋圖譜可對中藥的已知和未知成分進行全面綜合地表征,化學模式識別可以對指紋圖譜的信息進行數字化表達、識別和處理,兩者結合可以更加科學、客觀、系統地反映藥材的質量信息,并越來越多的用于中藥材的質量評價[14,15]。本研究擬采用高效、高靈敏度的UPLC方法,建立白芷不同產地和加工方式的UPLC指紋圖譜,采用對照品對各共有色譜峰進行化學指認,并結合聚類分析(cluster analysis,CA)、主成分分析(principalcomponent analysis,PCA)以及正交偏最小二乘法-判別分析(orthogonal partial least squares discriminant analysis,OPLS-DA)等化學模式識別方法對所得指紋圖譜進行綜合分析,為白芷藥材全面的質量控制提供理論依據。
1 儀器與試藥
1.1 儀器
Acquity UPLC H-Class超高效液相色譜系統,配備Acquity UPLC PDA檢測器、Empower2工作站(美國Waters公司);Legend Micro 17R微量離心機(美國Thermo公司);CPA225D電子天平(賽多利斯科學儀器有限公司);FA2004B電子天平(上海佑科儀器儀表有限公司);KQ-300DE型數控超聲波清洗器(昆山市超聲儀器有限公司);VORTEX-5渦旋儀(其林貝爾儀器制造有限公司)。
1.2 藥品與試劑
花椒毒酚(批號:HX106886198)、異紫花前胡內酯(批號:HM048527198)、水合氧化前胡素(批號:HA061413198)、白當歸素(批號:HB157470198)、補骨脂素(批號:HP018120198)、花椒毒素(批號:HA061509198)、佛手柑內酯(批號:HB204394198)、氧化前胡素(批號:HA062924198)、歐前胡素(批號:HI041232198)、異歐前胡素(批號:HI041233198),以上對照品均購自寶雞辰光生物科技有限公司,純度>98%。甲醇(色譜純,美國Fisher公司)、乙腈(色譜純,美國Fisher公司),其余為分析純;水為屈臣氏純凈水。本實驗搜集了四川、杭州、安徽、河南、河北等地的白芷藥材樣品,信息見表1,經陜西中醫藥大學陜西省中藥資源產業化協同創新中心劉世軍副教授鑒定,為白芷Angelicadahurica(Fisch.ex Hoffm.)Benth.et Hook.F或杭白芷A.dahurica(Fisch.ex Hoffm.)Benth.et Hook.F.var.formosana (Boiss.)Shan et Yuan的干燥根,其中S1、S3、S4、S7批次為硫磺熏蒸過的白芷樣品,S2、S5、S6、S8、S9、S10批次為無硫磺熏蒸過的白芷樣品。

表1 白芷藥材樣品信息
續表1(Continued Tab.1)

批次Batch編號Sample藥材Medicinalmaterial產地Origin加工方式ProcessingmethodS52018042205杭白芷浙江無硫熏S62018042206亳白芷安徽亳州無硫熏S72018042207亳白芷安徽亳州硫熏S82018042208禹白芷河南禹州無硫熏S92018042209祁白芷河北安國無硫熏S102018042210祁白芷河北安國無硫熏
2 方法與結果
2.1 色譜條件
色譜柱:ACQUITY UPLC BEH C18(50 mm×2.1 mm,1.7 μm);流動相:水(A)-乙腈(B);梯度洗脫(0~5 min,10%~25% B;5~18 min,25%~60% B;18~22 min,60%~90% B;22~25 min,90%~10% B;25~30 min,10% B);流速:0.2 mL/min;檢測波長:254 nm;進樣量為2 μL;柱溫25 ℃。在上述條件下,各色譜峰分離度良好,10批藥材的指紋圖譜和對照圖譜見圖1、圖2。
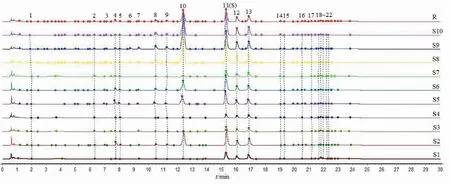
圖1 10批白芷藥材的UPLC指紋圖譜Fig.1 UPLC fingerprints of ten batches of A.dahurica 注:R 為對照指紋圖譜;S1~S10 分別為1~10批白芷藥材樣品。Note:R-the reference fingerprint;S1~S10 were A.dahurica samples.
2.2 對照品溶液的制備
精密稱取各對照品適量,分別加甲醇溶解并稀釋制備成濃度為1 mg/mL的對照品儲備溶液;精密吸取上述各對照品儲備溶液適量,混合后加甲醇稀釋,配置混合對照品儲備液,其中含歐前胡素、異歐前胡素和氧化前胡素500 μg/mL,水合氧化前胡素250 μg/mL,花椒毒酚、白當歸素和佛手柑內酯50 μg/mL,異紫花前胡內酯、補骨脂素和花椒毒素25 μg/mL。
2.3 供試品溶液制備
取白芷樣品粉末(過3號篩)約1 g,精密稱定,置于50 mL的具塞錐形瓶,加入甲醇20 mL,稱重,超聲(功率300 W,頻率50 kHz)提取1.5 h,取出,放冷,用甲醇補足重量,搖勻,濾過,即得供試品溶液,經0.22 μm濾膜濾過后進入UPLC分析。
3 指紋圖譜研究結果與分析
3.1 方法學考察
3.1.1 精密度試驗
按“2.3”項下方法制備1份供試品溶液,在“2.1”項下色譜條件重復進樣6次,由于歐前胡素在各批次白芷中均有出現,分離度良好且峰面積較大,故選擇歐前胡素(11號峰)作為參照峰(S),計算各共有峰的相對保留時間和相對峰面積。相對保留時間和峰面積RSD均在3%以內,表明儀器精密度良好。
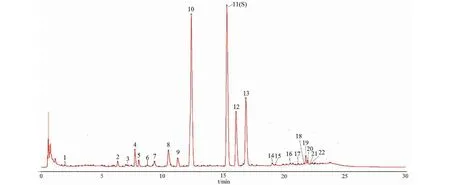
圖2 白芷藥材的對照圖譜Fig.2 Reference fingerprints of A.dahurica
3.1.2 穩定性試驗
按“2.3”項下方法制備供試品溶液,室溫下放置,按“2.1”項下色譜條件分別在0、2、4、8、12、24、48 h時進樣分析,以歐前胡素作為參照峰(S),計算各共有峰的相對保留時間和相對峰面積。相對保留時間和峰面積RSD均在3%以內,表明供試品溶液在48 h內穩定性良好。
3.1.3 重復性試驗
按“2.3”項下方法平行制備6份供試品溶液,“2.1”項下色譜條件進樣分析,以歐前胡素作為參照峰(S),計算各共有峰的相對保留時間和相對峰面積。相對保留時間和峰面積RSD均在3%以內,符合指紋圖譜測定要求。
3.2 指紋圖譜的建立及相似度評價
取10批白芷藥材按“2.3”項下方法制備供試品溶液,在“2.1”項色譜條件下檢測,記錄色譜圖。將所有數據導入國家藥典委員會“中藥色譜指紋圖譜相似度評價系統(2012.1版本)”軟件進行處理,標定22個共有峰,見圖1。10批白芷藥材指紋圖譜與對照指紋圖譜比對,進行相似度評價,其相似度依次為0.744、0.998、0.723、0.744、0.979、0.983、0.802、0.995、0.939、0.991,介于0.723~0.998之間。硫熏批次樣品(S1、S3、S4、S7)圖譜與對照圖譜的相似度范圍為0.723~0.802,小于 0.9;無硫熏批次樣品(S2、S5、S6、S8、S9、S10)與對照圖譜的相似度范圍為0.939~0.998, 均大于0.9,可見無硫熏藥材質量與對照圖譜比較接近,質量穩定,優于硫熏樣品。
3.3 主要色譜峰的化學指認
本研究采用對照品對各共有色譜峰進行化學指認,經比對保留時間和紫外光譜兩項指標,最終確認了2、3、4、5、6、7、8、10、11、13等10個色譜峰,分別為花椒毒酚、異紫花前胡內酯、水合氧化前胡素、白當歸素、補骨脂素、花椒毒素、佛手柑內酯、氧化前胡素、歐前胡素和異歐前胡素等呋喃香豆素成分(見圖3)。
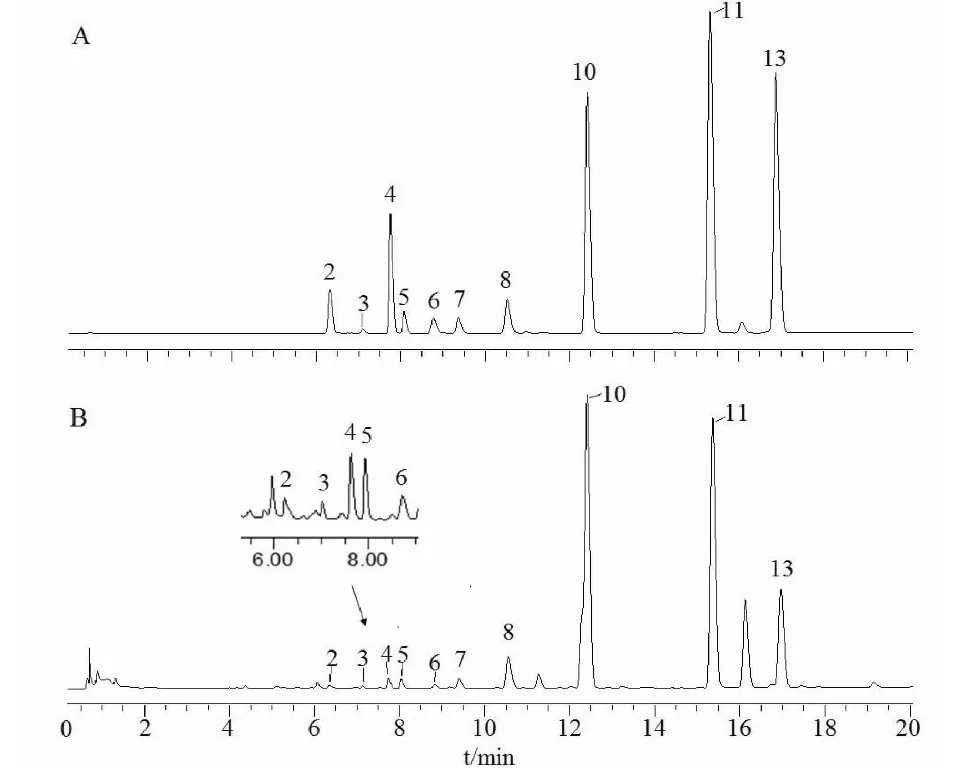
圖3 混合對照品(A)和白芷樣品(B)的UPLC色譜圖Fig.3 UPLC chromatograms of mixed standard (A) and A.dahurica (B)注:2-花椒毒酚;3-異紫花前胡內酯;4-水合氧化前胡素;5-白當歸素;6-補骨脂素;7-花椒毒素;8-佛手柑內酯;10-氧化前胡素;11-歐前胡素;13-異歐前胡素。Note:2-xanthotoxol;3- marmesin;4-oxypeucedaninhydrate;5-byak-angelicin;6-psoralen;7-xanthotoxin;8-bergapten;10-oxypeucedanin;11-imperatorin;13-isoimperatorin.
3.4 聚類分析(CA)
選擇白芷指紋圖譜中22個共有峰,將其峰面積相對于稱樣量量化,將10批樣品UPLC圖譜中22個共有峰的峰面積值標準化,組成10×22階原始數據矩陣,運用SPSS 22.0數據統計軟件,選用平均組間連接聚類,利用平均歐式距離法作為樣品間距離計算方法進行系統聚類分析,結果如圖4所示,橫坐標為樣品間的距離,縱坐標為白芷藥材樣品編號(與表1相同)。由圖4可知,各批次白芷樣品主要分為兩大類,S1~S7聚為一大類(Ⅰ組),其中S1、S3、S4、S7均為硫熏批次樣品,聚為一類;S2、S5、S6分別為四川、杭州、安徽無硫熏批次樣品,聚為一類;S8、S9、S10為河南、河北產地無硫熏批次樣品,聚為一大類(Ⅱ組)。
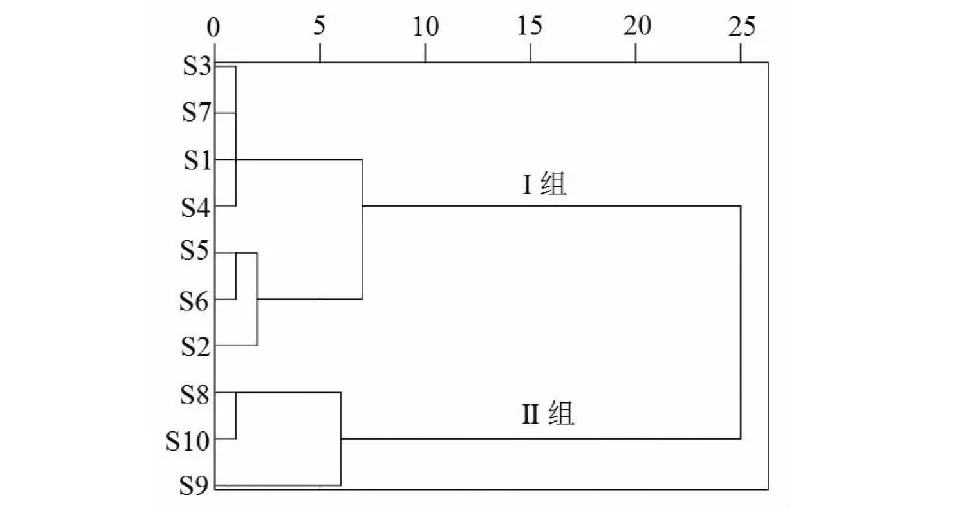
圖4 聚類分析結果Fig.4 Results of hierarchical cluster analysis
3.5 主成分分析(PCA)
用SPSS 22.0軟件對量化的各共有峰峰面積進行標準化處理后,對10批白芷藥材指紋圖譜所得的22個共有峰進行主成分分析,求出相關矩陣的特征值及其方差,見表2。共提取出4個主成分,其中前3個主成分的累積方差貢獻率達到81.7%,可代表白芷指紋圖譜共有峰的大部分信息。
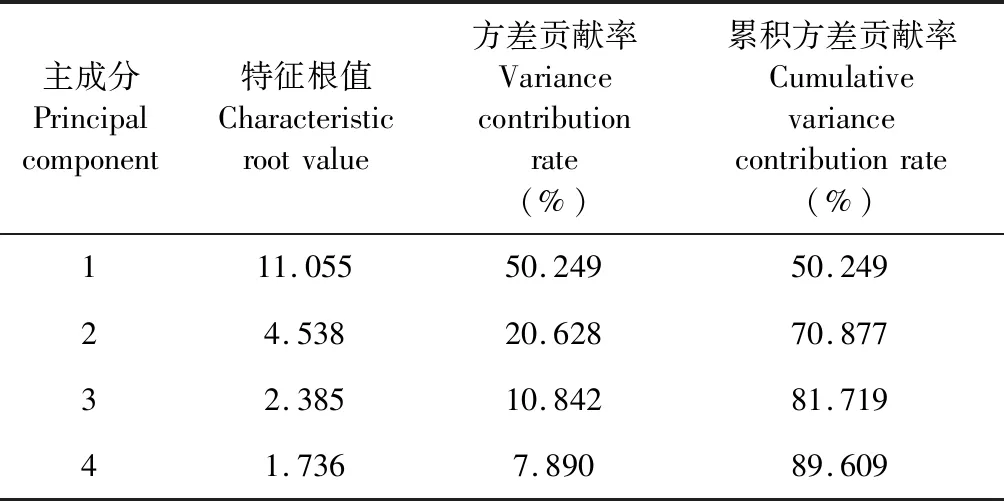
表2 主成分特征值及方差
主成分載荷矩陣反映了各變量對主成分的貢獻大小和作用方向,由圖5可見,22個變量對主成分1多成正相關,其中共有峰3、8~14、16、18、21對其貢獻最大;共有峰15、19、20對主成分2貢獻最大,共有峰4、5與其呈負相關;共有峰4、15、17對主成分3貢獻最大,共有峰7與其呈負相關。
進一步通過將各特征向量中心化和標準化后,10批樣品主成分得分如圖6所示,并以樣品的第 1、2、3主成分得分做三維散點圖,見圖7。如圖所示,S1、S3、S4、S7硫熏批次樣品聚為一類,S2、S5、S6批次樣品聚為一類,這與聚類分析結果完全一致;S8、S9批次樣品聚為一類,S10號無硫熏批次樣品,雖與S8、S9距離稍遠,但均與第1主成分成正相關。
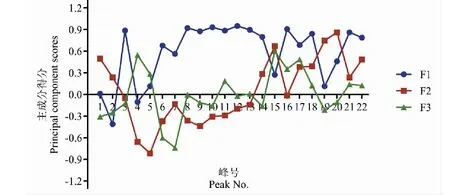
圖5 22個共有峰3個主成分的排序坐標圖Fig.5 Coordinate diagram of three principal components in 22 common peaks
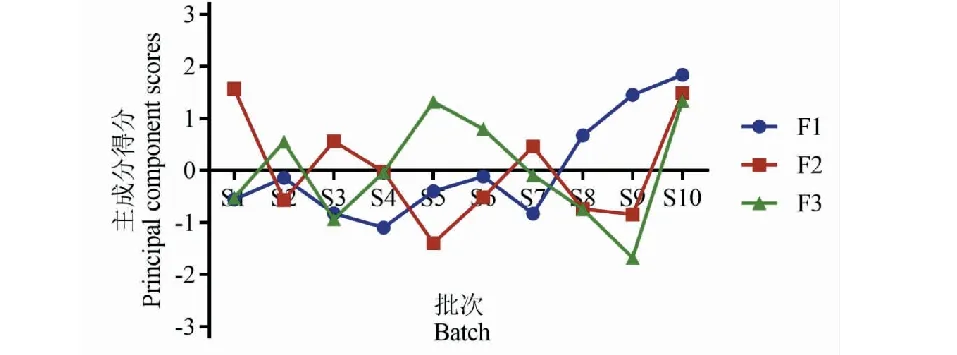
圖6 10批白芷藥材3個主成分的排序坐標圖Fig.6 Coordinate diagram of three principal components in ten batches
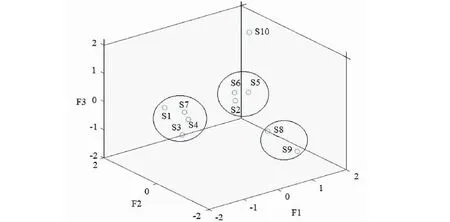
圖7 主成分得分圖Fig.7 PCA score figure
3.6 正交偏最小二乘法-判別分析(OPLS-DA)
為進一步篩選出對不同批次白芷藥材質量貢獻較大的成分,本研究采用Simca 14.1軟件進行OPLS-DA分析。篩選變量重要性投影值(variable importance in project,VIP)>1.0的色譜峰,即該色譜峰對整體模型的貢獻度高于平均水平。結果見圖8,橫坐標為指紋圖對應的色譜峰編號,縱坐標為各色譜峰對應的VIP值,根據VIP>1.0,共找到8個有意義變量,按照VIP值大小依次為 11號峰(歐前胡素)、8號峰(佛手柑內酯)、9號峰、12號峰、10號峰(氧化前胡素)、13號峰(異歐前胡素)、16號峰、5號峰(白當歸素)。表明上述色譜峰所對應的化合物是白芷各批次藥材之間產生差異的主要標志性成分,提示在白芷的質量控制和品質評價中,應重點關注這些成分的質量變化。
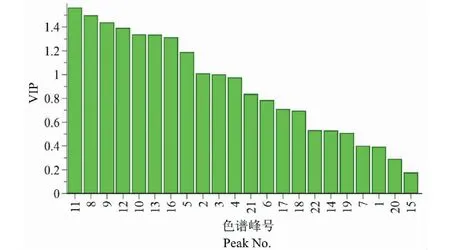
圖8 10批次白芷藥材中各活性成分的VIP值Fig.8 VIP plot of active ingredients in ten batches of A.dahurica
3.7 組間差異性分析
為進一步分析影響藥材質量差異的主要成分,對22個共有峰進行有無硫熏以及不同產地的組間差異性分析,結果見表3。結果顯示,22個共有峰中有8個峰的峰面積在硫熏批次樣品(S1、S3、S4、S7)和無硫熏批次樣品(S2、S5、S6、S8、S9、S10)間存在顯著差異(P<0.05),無硫熏樣品中差異峰的峰面積顯著高于硫熏樣品;8個差異峰分別是5號峰、8~13號峰和16號峰,與OPLS-DA分析結果是一致的。
由于收集的相同產地批次樣品較少,根據聚類分析結果以及白芷產地的地理位置,分析北方產地(河南、河北)和南方產地(四川、浙江、安徽)無硫熏批次樣品間的差異性成分。組間差異性結果(表3)顯示: 4號峰、8~13號峰和14號峰的峰面積在北方和南方產地中均存在顯著差異(P<0.05),除4號峰外,其他差異峰在北方產地的峰面積顯著高于南方產地。

表3 組間差異性分析
注:*P<0.05,**P<0.01,***P<0.001 無硫熏批次樣品vs硫熏批次樣品;#P<0.05,##P<0.01,###P<0.001 北方產地vs南方產地。
Note:*P<0.05,**P<0.01,***P<0.001 samples without sulfur fumigationvssamples with sulfur fumigation;#P<0.05,##P<0.01,###P<0.001 the northern originvsthe southern region.
4 結論
本研究分別對提取溶劑(50%甲醇、75%甲醇、純甲醇、50%乙醇、75%乙醇、乙醇)、藥材-溶液比(1∶10、1∶20、1∶50、1∶100)以及超聲提取時間(0.5、0.75、1.0、1.5 h)進行了系統考察,以提取方法穩定可行,提取效率最優,色譜峰響應、分離度良好為指標,最終選擇的提取溶劑為純甲醇、藥材-溶液比為1∶20、超聲提取時間為1.5 h。
通過考察了樣品在甲醇-水、乙睛-水、甲醇-酸水和乙睛-酸水作為流動相系統時,各色譜峰的分離效果,發現乙睛-水作為流動相系統時,各色譜峰的分離度最好,峰形最佳,基線較平穩。用 PDA 檢測器對樣品進行190~400 nm全波長掃描,比較各波長下色譜圖:除異紫花前胡內酯在325 nm左右有較大吸收外,其他呋喃香豆素成分均在254 nm有較大吸收,且在254 nm處,色譜峰個數較多,分離度好,響應合理,因此選定254 nm為白芷藥材指紋圖譜的測定波長。
白芷是一味重要的傳統中藥,臨床應用廣泛,但對其質量控制,尚缺乏系統的研究報道。本研究選取不同產地及處理方式的白芷藥材作為研究對象,建立了UPLC指紋圖譜,共標定了22個共有峰,通過對照品對其中的10個成分進行化學指認。依次運用相似度分析、CA、PCA、OPLS-DA、組間差異性分析這5種方法對不同產地的白芷藥材 UPLC指紋圖譜進行分析,相似度分析、CA和PCA結果均說明,硫磺熏蒸是導致白芷藥材質量差異的重要因素,沒有硫磺熏蒸的白芷藥材質量明顯優于硫磺熏蒸樣品。通過CA和PCA分析,硫熏批次樣品聚為一類,四川、杭州、安徽無硫熏批次樣品聚為一類,河南河北產地無硫熏批次樣品聚為一類,說明各產地白芷藥材質量存在一定差異。進一步采用OPLS-DA,篩選出影響不同批次白芷藥材質量的主要成分,共找到8個主要差異性成分,經對照品化學指認出其中5個成分,分別為歐前胡素、佛手柑內酯、氧化前胡素、異歐前胡素和白當歸素,還需進一步借助質譜等方法對其他3個重要的差異質量標志物進行鑒定分析。最后對22個共有峰進行了有無硫熏以及不同產地的組間差異性分析,有無硫熏兩種不同處理方式間有顯著差異的成分與OPLS-DA分析的主要成分相同,無硫熏樣品優于硫熏樣品,這與硫熏影響白芷的鎮痛作用研究是相符的,為硫熏導致白芷的藥效作用降低提供了物質基礎;北方省份(河南、河北)和南方省份(四川、浙江、安徽)的白芷樣品間也存在較顯著差異,河南河北產地樣品優于四川、浙江和安徽產地樣品。8~13號共有峰是影響白芷藥材質量的中藥成分,雖未見不同產地白芷藥材藥效作用差異的研究報道,但根據我們前期的藥代動力學研究結果[1,3],8號峰(佛手柑內酯)、10號峰(氧化前胡素)、11號峰(歐前胡素)、13號峰(異歐前胡素)等均是白芷藥材的主要入血成分,且這些成分也是影響白芷藥效的主要成分,其含量高低直接影響臨床療效。上述實驗說明,硫熏和地理位置是影響白芷藥材質量的兩個重要因素,可通過主要差異性成分對不同處理方式和產地的白芷藥材質量進行分析。
本研究首次采用指紋圖譜結合化學模式識別技術,較為系統、整體和全面的對不同批次白芷藥材質量進行了深入探索,對不同產地和處理方法的白芷藥材進行分類,并篩選出了影響白芷藥材質量的差異質量標志物,為白芷活性成分的鑒定、分析、質量控制和物質基礎研究等提供了準確、豐富的信息,補充了現有質量評價方法的不足,同時為中藥全面質量評價研究提供借鑒與思路。
