H2O2在α-Fe2O3表面非均相氧化SO2機理的模擬研究
2019-11-28 05:15:08李冬坤董發勤李海龍霍婷婷賀小春趙玉連
巖石礦物學雜志 2019年6期
關鍵詞:結構
李冬坤,董發勤,李海龍,霍婷婷,賀小春,趙玉連,彭 潔
(1. 西南科技大學 環境與資源學院, 四川 綿陽 621010; 2. 固體廢物處理與資源化教育部重點實驗室,四川 綿陽 621010; 3. 西南科技大學 材料科學與工程學院, 四川 綿陽 621010)

過氧化氫(H2O2)是一種重要的大氣氧化劑,與對流層中HOx自由基收支和氧化能力直接相關。在實際大氣中,礦物氣溶膠上的非均相反應是SO2、氣態H2O2重要的匯(Reeves and Penkett, 2003; Pradhanetal., 2010; Zhaoetal., 2013)。Song和Zhao等結合計算模擬和實驗研究證實了H2O2能夠在過渡金屬氧化物、礦物粉塵表面分解產生·OH自由基(Zhaoetal., 2014; Songetal., 2017, 2018)。·OH自由基的存在更有利于礦物氣溶膠上的非均相氧化的發生(Bouya, 2015)。此外,Huang等發現H2O2的存在可以提高SO2在礦物粉塵上的吸附率——在有H2O2存在的情況下,SO2的飽和覆蓋率比無H2O2存在的情況下大5倍左右。另外,在有H2O2存在的干燥條件下,真實礦塵對SO2的吸收系數約提高30%~50%(Huangetal., 2015, 2016)。然而,目前關于H2O2的這種促進作用及H2O2在過渡金屬氧化物α-Fe2O3上氧化SO2的非均相反應機理信息仍知之甚少,SO2和H2O2共存時在α-Fe2O3上的優先吸附順序以及SO2是如何與礦物氧化物結合并發生轉化的途徑仍不夠清楚,需要做進一步的研究。
因此,本文選取大氣中無機氣體(SO2)、H2O2和赤鐵礦(α-Fe2O3)作為研究對象,采用巨正則蒙特卡羅方法(Grand Canonical Monte Carlo, GCMC)和密度泛函理論,模擬計算了SO2、H2O2在α-Fe2O3(001)表面不同吸附構型的吸附和非均相氧化作用,進一步分析SO2、H2O2在α-Fe2O3上的非均相氧化機制,為更好地評估SO2、H2O2非均相反應對大氣中礦物氧化物老化作用提供重要信息。
1 計算細節
1.1 α-Fe2O3晶體結構模型建立
選用具有最穩定結構的鐵氧化物赤鐵礦(α-Fe2O3)為對象,建立α-Fe2O3晶體結構模型。該構型主要基于氧原子的六方密排晶格,具有R-3C立方空間群結構。其中Fe原子僅僅占據了氧原子之間的2/3的氧八面體空位,如圖1所示。經過優化后的結構參數(a=b=5.03 ?,c=13.77 ?;α=β= 90°,γ=120°)與實驗報道的結構參數(a=b=5.04 ?,c=13.75 ?;α=β=90°,γ=120°)較一致(Songetal., 2013)。Weiss和Song等人的理論計算研究發現包含Fe—O3—Fe端面的α-Fe2O3(001)晶面是主要的自然生長晶面,其催化活性高于其它晶面(Weissetal., 2002; Songetal., 2017),因此本文主要討論α-Fe2O3(001)晶面與SO2間的非均相反應。為了研究α-Fe2O3與SO2的相互作用,所有結構模型均采用了包含4個α-Fe2O3原胞結構的周期性重復的超晶胞(2×2×1),且在z軸方向建立了16 ?的真空層,并考慮了α-Fe2O3(001)表面上不同的吸附位點(Fe位和O位)和吸附分子不同的吸附取向對吸附機制的影響。
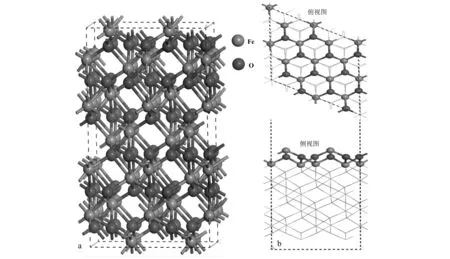
圖 1 α-Fe2O3晶體結構圖(a)和α-Fe2O3(001)表面的俯視圖和側視圖(b)Fig. 1 Crystal structure diagram of α-Fe2O3 (a), top and side views of the surface of α-Fe2O3 (001) (b)
1.2 計算方法
本文中涉及的計算均借助于美國Accelrys公司提供的Material Studio軟件。首先,采用GCMC方法將結構優化后的SO2、H2O2分子吸附在α-Fe2O3(001)表面,通過考慮不同的吸附位點(Fe位和O位),建立不同的吸附結構模型并對吸附體系的相互作用能進行計算。其次,在不同的系宗(NVE和NPT)中采用共軛梯度的方法,利用Castep模塊中的Geometry Optimization和Dynamic對不同的吸附結構模型進行結構優化和分子動力學馳豫,保證不同的吸附結構模型達到平衡態。其中,短程的范德華力和長程的靜電相互作用分別用Atom-Based和Ewald+Group方法進行模擬,溫度和壓力分別由Nose-Hoover thermostat和Berendsen-Barostat方法控制。最后,在相同的計算模塊(Castep模塊)中,采用基于廣義梯度近似(Generalized Gradient Approximation,GGA)的PBE(Perdew-Burke-Ernzerhof)交換關聯泛函和投影綴加平面波法(Augmented-Plane-Wave,APW)計算了體系的電子結構。在考慮強關聯體系中的高度局域化Fe-3d軌道作用的前提下,本文采用超軟贗勢(Ultrasoft Pseudopotentials)和GGA+U的方法計算Fe-3d軌道能級結構(Bianetal.,2017)。其中,Castep模塊參數為: Monkhorst-Pack的布里淵區為3×3×3,GGA+PBE描述電子間的交換關聯函數,超軟贗勢描述離子實與價電子之間的相互作用。自洽場運算中采用Pulay密度混合法,平面波截止能為300 eV,收斂精度SCF為2×10-5eV/atom。對于H2O2和SO2在α-Fe2O3(001)表面上的吸附能計算依賴于公式:
E=ET- (EF+Es)
式中的ET指H2O2或者SO2吸附在α-Fe2O3表面上的總能,EF指孤立的H2O2或者SO2分子的總能,ES指獨立的α-Fe2O3(001)表面的總能。負的吸附能表明H2O2或者SO2分子在α-Fe2O3(001)表面的吸附是放熱反應,負的吸附能的絕對值越大,表明分子與表面的相互作用越強。
2 結果與討論
2.1 SO2在α-Fe2O3 (001)表面的吸附
SO2表面吸附是SO2在α-Fe2O3(001)上發生非均相反應的重要步驟。考慮到不同的吸附位點,對SO2在α-Fe2O3(001)表面上不同吸附位點吸附的結構進行了優化,得到兩種穩定的吸附構型1A(Fe—S)和1B(Fe—O),如圖2所示。其中,1A和1B構型是由SO2分別通過S原子和O原子與表面的Fe原子進行吸附,相應吸附構型1A的結構參數為: Fe—S=2.43 ?,Fe—O=1.73 ?;吸附構型1B的結構參數為: Fe—O=1.93 ?; S—O=1.72 ?。吸附能如表1所示。

圖 2 SO2在α-Fe2O3(001)表面的兩種吸附構型Fig. 2 Two adsorption configurations of SO2 on the surface of α-Fe2O3 (001)

表 1 不同吸附構型的吸附能 E/eVTable 1 Adsorption energy of different adsorption configurations
由表1可以看出,吸附構型1A的吸附能(-0.17 eV)明顯小于吸附構型1B的吸附能(2.90 eV),由此說明SO2在α-Fe2O3(001)表面的化學吸附是通過SO2的S原子和α-Fe2O3的Fe原子發生的。SO2分子的S—O鍵也從1.54 ?增大到1.73 ?,說明SO2分子在α-Fe2O3表面吸附后被激活,S原子與Fe原子間的鍵長為2.43 ?。
為深入解析SO2在α-Fe2O3(001)表面的吸附機制,計算了1A和1B的能帶結構和局域態密度(PDOS)。圖3表明,在吸附構型1A中的Fe-3d和O-2p軌道在能量區域內(-2.0 ~ 0.5 eV)、(-6.4~-3.2 eV)和(-11.8 ~-10.3 eV)有明顯的重疊;在吸附構型1B中的Fe-3d和S-2p軌道在(-7.2~0.5 eV)和(-12.0 ~-10.6 eV)有明顯的重疊。但是Fe-3d與S-2p軌道的重疊程度明顯高于Fe-3d與O-2p軌道重疊程度,這表明SO2在α-Fe2O3(001)表面的吸附更傾向于通過d-p(Fe-3d-S-2p)軌道雜化作用,這一結果不僅與前面計算的吸附能結果一致,而且與Baltrusaitis等報道SO2在α-Fe2O3表面吸附的實驗結果一致(Baltrusaitietal., 2007)。為進一步說明相互作用機制,計算了電荷密度和前線軌道說明吸附體系中電子的轉移,結果如圖3b和3c所示。由前線軌道可知SO2與α-Fe2O3(001)間的電子轉移主要依賴于Fe原子和S原子之間,同時根據差分電荷密度和總電荷密度發現在Fe原子和S原子之間有明顯的電子轉移,由此說明Fe原子和S原子間的d-p(Fe-3d-S-2p)軌道雜化是SO2在α-Fe2O3(001)表面吸附的主要機制。

圖 3 α-Fe2O3+ SO2不同吸附構型的PDOS圖(a)、表面電荷密度圖(b)和前線軌道圖(c)Fig. 3 PDOS charts of different adsorption configurations of α-Fe2O3 + SO2 (a), surface charge density (b) and frontier orbital maps (c)
以Mulliken電荷分析作為定量分析電子轉移的重要依據, 由表2中所示的Fe原子和S(或O)原子Mulliken電荷表明電子的轉移是從α-Fe2O3表面向SO2轉移, 吸附構型1A和1B的電子轉移分別為0.61 e和0.43 e,說明吸附構型2A的相互作用強于吸附構型2B,這一結論與吸附能和能帶結構分析結果一致。因此,α-Fe2O3(001)表面的Fe作為吸附SO2的活性位點,吸附構型2A作為SO2在α-Fe2O3(001)表面吸附的最優吸附方式,d-p(Fe-3d-S-2p)軌道雜化是Fe—S鍵形成的主要原因。
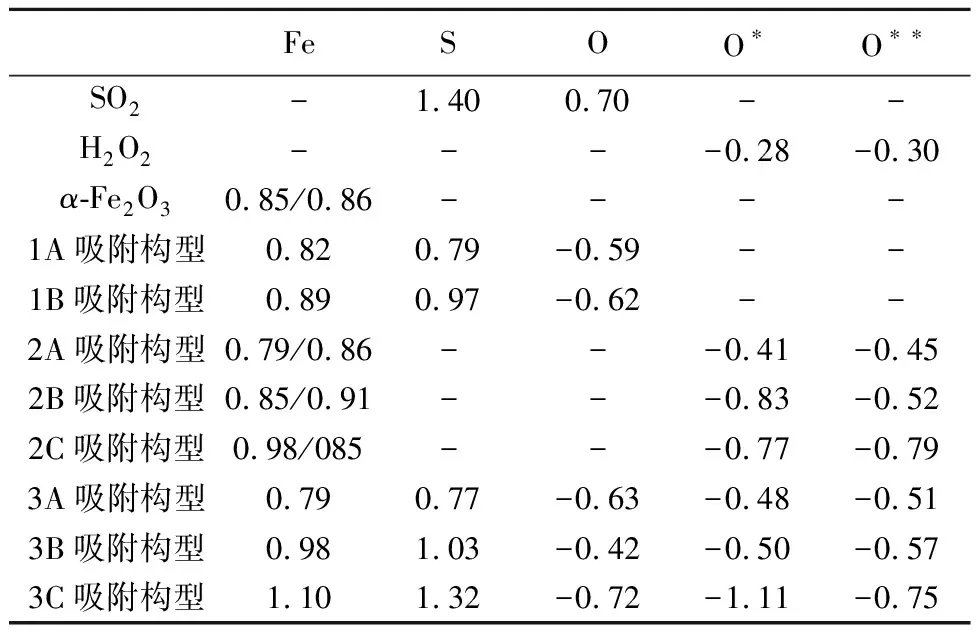
表 2 不同原子電荷Mulliken布局分析 eTable 2 Mulliken layout analysis of different atomic charges
注: O代表SO2中的O,O*和O**分別代表H2O2中的兩個O。
2.2 H2O2在α-Fe2O3(001)表面的吸附與分解
H2O2能夠在礦物氧化物上發生非均相分解產生·OH,具有氧化性的·OH基團氧化作用是氧化SO2的重要途徑,H2O2在α-Fe2O3表面的分解可作為氧化性·OH基團產生的主要來源。因此,研究H2O2在α-Fe2O3(001)表面的吸附機制,可以為研究·OH基團氧化SO2提供依據。考慮到不同的吸附位點,得到了H2O2在α-Fe2O3(001)表面的吸附構型2A與分解吸附構型2B和2C。其中,吸附構型2A是H2O2分子通過O原子與表面的Fe原子吸附;分解吸附構型2B和2C是由H2O2分子分別分解為H2O+O和·OH+·OH與表面的Fe原子吸附,如圖4所示。在吸附構型2A中,H2O2通過表面的鐵原子分子平行吸附在α-Fe2O3(001)表面。H2O2與α-Fe2O3(001)表面不僅形成Fe—O鍵(1.95 ?),而且H2O2的兩個氫原子與兩個表面氧原子形成兩個氫鍵的長度分別為2.89 ?和2.70 ?。與SO2的吸附相似,表面的Fe原子作為H2O2吸附的活性位點,H2O2的吸附是通過H2O2的O原子和表面的Fe原子。同時,在α-Fe2O3(001)表面吸附H2O2的O—O鍵相比于獨立的H2O2明顯增強(1.54 ?→2.85 ?)。為此,考慮了H2O2以不同的分解方式在α-Fe2O3(001)表面的吸附,得到了穩定的吸附構型2B和2C。吸附能計算表明吸附構型2C相比于吸附構型2A和2B具有較低的吸附能,由此說明H2O2在α-Fe2O3(001)表面的吸附方式是以兩個·OH基團與和α-Fe2O3的Fe原子結合,并以此作為最佳的吸附構型。
為進一步描述H2O2與α-Fe2O3(001)表面間的吸附和分解機制,我們計算了吸附體系的能帶結構和電荷密度。由圖5a所示,能帶結構(-20.0~0.8 eV)是由Fe-3d、O-2p軌道構成。對比發現在2A、2B和2C中H2O2的O-2p軌道發生了明顯的變化,其中在吸附構型2C中O-2p軌道相比于吸附構型2A和2B發生了明顯的軌道偏移和簡并。O-2p軌道偏移和簡并[-11.3~-1.6 eV (2A)和-8.4~-3.8 eV (2B)→-6.7~0.7 eV (2C)]增大了Fe-3d與·OH基團的O-2p軌道的重疊程度,增強了d-p(Fe-3d-O-2p)軌道雜化作用。同時,由差分電荷密度圖和總電荷密度圖可以看出羥基·OH基團的氧原子與羥基結合的兩個鐵原子間有明顯的電荷轉移積累。這一現象表明,電荷密度從Fe原子轉移到了羥基的O原子,·OH與α-Fe2O3(001)表面發生了相互作用。由于在Fe2O3中只有2/3的八面體被Fe原子占據,這使得表面部分的Fe原子處于高電子組態。根據晶體場理論和能量最小原則,dz2和dx2+y2相比于dxy、dyz和dxz軌道指向配體,這使得Fe-3d與H2O2的O-2p軌道間的d-p(Fe-3dz2+O-2px和Fe-3dz2+O-2py)雜化方式轉變為Fe-3d與·OH基團的O-2p軌道間的d-p(Fe-3dz2+O-2py)雜化方式。通過Mulliken電荷分析發現電子的轉移是從α-Fe2O3表面向H2O2轉移,吸附構型2A、2B和2C的電子轉移分別為0.14 e、0.39 e和0.49 e。這說明吸附構型2C的相互作用強于吸附構型2A和2B。因此,Fe作為吸附H2O2的活性位點,吸附構型2C作為H2O2在α-Fe2O3(001)表面吸附的最優吸附方式,O-2p軌道的偏移和簡并是增強d-p(Fe-3d-O-2p)軌道雜化的原因,也是吸附構型2C作為最優吸附方式的主要原因。

圖 4 H2O2在α-Fe2O3(001)表面的吸附與分解構型Fig. 4 Adsorption and decomposition configuration of hydrogen dioxide on the surface of α-Fe2O3 (001)

圖 5 H2O2在α-Fe2O3 (001)表面吸附時不同吸附構型的PDOS圖(a)、表面電荷密度(b)和前線軌道圖(c)Fig. 5 PDOS diagrams of different adsorption configurations of hydrogen dioxide on the surface of α-Fe2O3 (001) (a), surface charge density (b) and frontier orbital maps (c)
2.3 SO2和H2O2在α-Fe2O3 (001)表面共吸附
已有研究表明,SO2在α-Fe2O3(001)表面可以被H2O2氧化(Huangetal., 2015)。然而,僅僅計算H2O2在α-Fe2O3(001)表面吸附時,不能直接證實·OH基團氧化性的存在,因此,又研究了SO2和H2O2在α-Fe2O3(001)表面的共吸附作用機制。考慮SO2和H2O2的競爭吸附和H2O2在表面的分解,分別得到了3A、3B和3C共吸附構型,如圖6所示。在3A和3B中,SO2和H2O2分子被吸附到α-Fe2O3(001)表面的Fe原子。根據吸附構型3A(-3.38 eV)和3B(-2.29 eV),SO2和H2O2在α-Fe2O3(001)表面之間存在競爭性吸附,SO2分子相比于H2O2分子會優先吸附在α-Fe2O3(001)表面。在吸附構型3C中,H2O2在α-Fe2O3(001)表面分解形成兩個表面·OH基團,其中一個·OH基團通過氫鍵與SO2反應形成·OH+SO2團簇分子,鐵氧鍵Fe—O和氫鍵H—O的鍵長分別為1.91 ?和1.76 ?。同時,對比于SO2和H2O2共吸附時的吸附能,當H2O2分解為兩個表面·OH基團與SO2在α-Fe2O3(001)表面具有較低的吸附能,說明SO2和H2O2在α-Fe2O3(001)表面是通過H2O2產生的·OH基團將SO2氧化,以·OH+SO2團簇分子的形式吸附在α-Fe2O3(001)表面。這與Huang等實驗觀察到H2O2與SO2在礦物粉塵上迅速反應,H2O2在礦物粉塵上的非均相分解產生的·OH自由基會立即與吸附的SO2反應相一致(Huangetal., 2015)。

圖 6 SO2和H2O2在α-Fe2O3(001)表面共吸附構型Fig. 6 Co-adsorption configuration of SO2 and H2O2 on α-Fe2O3 (001) surface
為進一步探討SO2和H2O2在α-Fe2O3(001)表面的共吸附機制和·OH對SO2的氧化機制,我們計算了吸附構型3C的能帶結構和電荷密度。如圖7a所示,能帶結構的價帶(-20.0~0.8 eV)主要由Fe-3d、O-2p、S-2p軌道構成。對比發現在2·OH+α-Fe2O3(001)的O-2p4軌道H2O2+α-Fe2O3(001)發生了明顯的劈裂,這增大了Fe-3d與·OH的O-2p軌道的重疊程度,改變了原來的d-p(Fe-3d-O-2p)軌道雜化強度。同時,在吸附構型3C中·OH基團的O-2p軌道與SO2的S-2p軌道在費米面(-7.5~0.8 eV)附近的重疊程度也明顯增大,使得O-2p與S-2p軌道p-p(O-2p-S-2p)軌道雜化增強。差分電荷密度和總電荷密度計算表明,在構型3C中O和Fe原子(O—Fe鍵)與O和S原子(O—S鍵)周圍有明顯的電荷積聚,表明這些粒子之間有很強的相互作用。電子從表面Fe原子轉移到·OH的O原子,形成一個O—Fe鍵;在·OH+SO2團簇分子中,由于高度局域化的O-2p軌道具有較高的電負性,誘導處于半滿狀態的S-2p軌道電子轉移到另一個·OH的O原子的O-2p,形成一個S—O鍵,使得SO2被氧化[S(SO2)-電荷布局:0.79 e→1.32 e;O(H2O2)-電荷布局: -0.77 e→-1.11 e]形成·OH +SO2團簇。為進一步說明相互作用機制,進行了電荷的布局分析。通過Mulliken電荷分析發現電子的轉移是從α-Fe2O3表面向H2O2分解產生的·OH轉移,由SO2向H2O2分解得到的另一個·OH轉移,其電荷轉移(0.22 e)明顯強于吸附構型3A和3B的電子轉移(分別為0.07 e和0.10 e)。這表明吸附構型3C的相互作用強于吸附構型3A和3B。因此,吸附構型3C作為H2O2在α-Fe2O3(001)表面吸附的最優吸附方式。d-p(Fe-3d-O-2p)和p-p(S-2p-O-2p)軌道雜化方式的轉變是H2O2分解的主要原因。
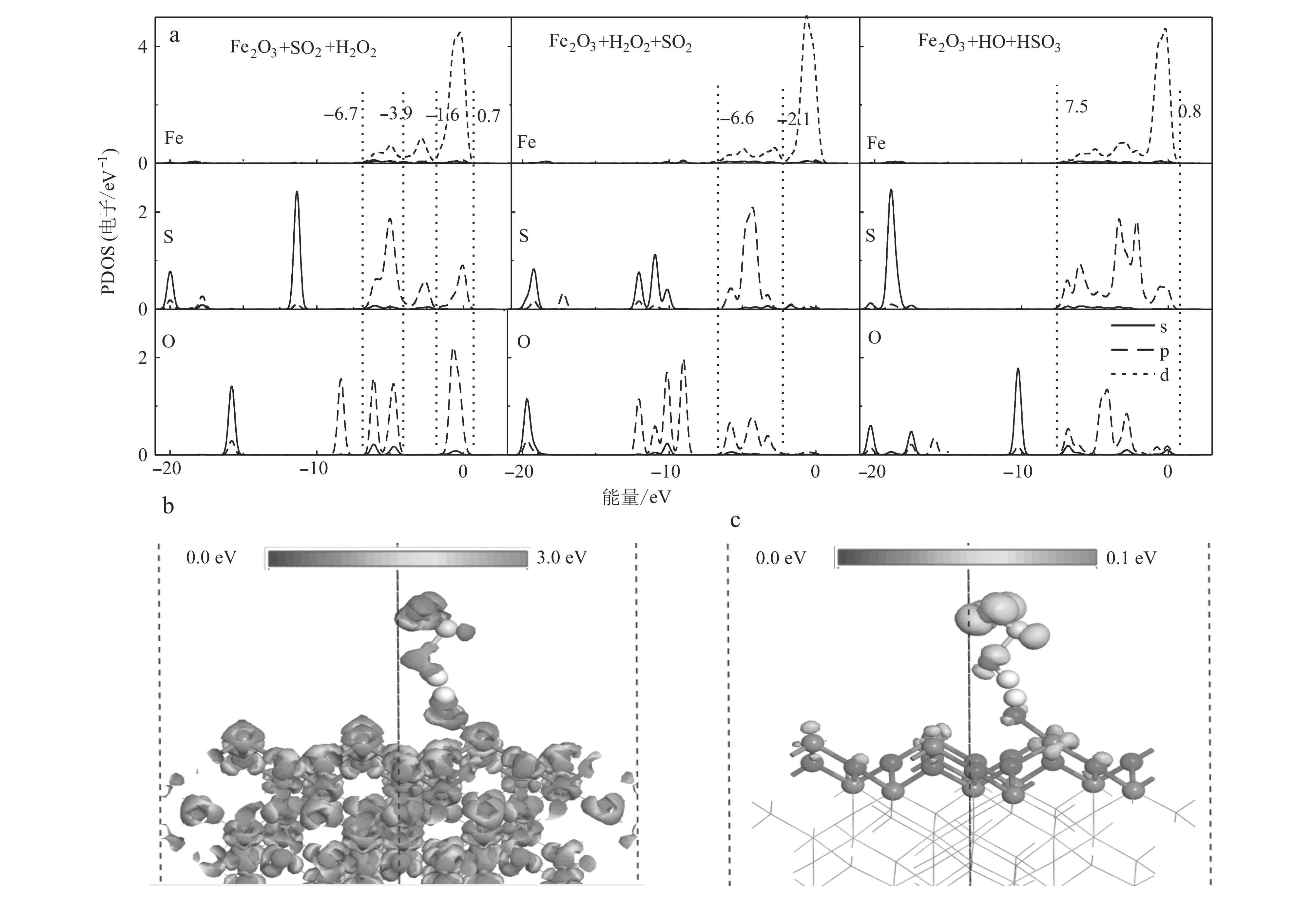
圖 7 SO2和H2O2在α-Fe2O3(001)表面共吸附的不同吸附構型的PDOS圖(a)、表面電荷密度圖(b)和前線軌道圖(c)Fig. 7 PDOS diagrams of different adsorption configurations for co-adsorption of SO2 and H2O2 on the surface of α-Fe2O3 (001) (a) , surface charge density (b) and frontier orbital (c) diagrams
3 結論
通過模擬計算SO2與H2O2在α-Fe2O3(001)表面的吸附機制,闡釋了SO2在α-Fe2O3(001)表面氧化機制。計算結果表明,SO2、H2O2的吸附分別是通過SO2的S原子、H2O2的O原子與α-Fe2O3(001)表面的Fe原子間的d-p(SO2: Fe-3d-S-2p;H2O2: Fe-3d-O-2p)軌道發生雜化作用。特別地,H2O2在α-Fe2O3(001)表面的最優賦存形式是以兩個·OH基團與α-Fe2O3(001)表面的Fe原子吸附。當SO2與H2O2在α-Fe2O3(001)表面共吸附時,H2O2分子以·OH基團優先吸附在α-Fe2O3(001)表面,一個·OH基團與吸附的SO2發生氧化反應,將SO2氧化為·OH+SO2團簇分子[S(SO2)-電荷布局: 0.79 e→1.32 e;O(H2O2)-電荷布局: -0.77 e→-1.11 e]。·OH+SO2團簇分子與另一個·OH基團通過氫鍵吸附在α-Fe2O3(001)表面。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
