多價抗蟲水稻外源Bt 基因的多引物多重PCR 檢測方法
2019-12-04 09:30:02方欣怡陽菁王井章李陽生
生物技術通報 2019年12期
方欣怡 陽菁 王井章 李陽生
(武漢大學生命科學學院 水稻國家重點實驗室,武漢 430072)
水稻鱗翅目害蟲會影響水稻豐產和穩產。稻縱卷葉螟、二化螟、三化螟對我國水稻生產造成的損失可占總產量的5%-10%,雜交水稻水稻的廣泛種植使得長江流域的三化螟危害下降,二化螟進而變成主要害蟲[1-2]。其主要危害表現為造成葉片枯白、枯心苗、白穗。目前在水稻中尚未發現對水稻鱗翅目害蟲具有抗性的稻種資源,因此過去對螟蟲的防治主要以施用農藥為主,但也存在防治效果不佳、農藥殘毒污染、增加成本等問題。
近10 年,我國政府大力支持轉Bt基因在糧食作物中的應用,投入了大量的人力、財力、物力。Bt基因是來自于蘇云芽孢桿菌的一種殺蟲基因,這種轉基因水稻可在體內表達出一種Bt 殺蟲蛋白,可特異性毒殺鱗翅目、鞘翅目昆蟲,減少殺蟲劑的使用量,是目前農作物轉基因育種主要使用的目標抗蟲基因[3-4]。在抗蟲育種及生產中發現,隨著Bt 殺蟲劑的大量應用,會使害蟲產生抗性,作用難以持久,而使其應用具有一定的風險。不同的Bt 蛋白在昆蟲體內的受體并不相同,同時利用多個Bt基因的多價抗蟲水稻,可以獲得抗蟲性狀的持久利用[5-7],而且不會明顯改變農藝性狀,單拷貝插入的后代分離符合孟德爾遺傳規律。目前國內水稻科學家們已經培育出一系列的轉Bt基因抗蟲水稻,如轉基因水稻TT51-1(轉cry1Ab/cry1Ac融合基因),轉基因水稻T2A-1(轉cry2A基因),轉基因水稻T1C-19(轉cry1C基因)等[4,5,8-10],并利用這些轉化事件通過雜交和系統選育的方法,培育了一批具有商業生產潛力的轉基因水稻新品系。這些抗蟲新品系不僅可以用來作為新的抗性育種資源,也可以用于發展三價Bt水稻[4]。利用多個Bt基因培育多價抗蟲水稻獲得持久的抗蟲性狀是抗蟲轉基因育種的發展趨勢。
選育Bt基因純合的優良單株是決定育種進程的一個關鍵因素。目前常用的檢測Bt基因的分子標記主要是根據外源插入片段序列設計的特異顯性標記,該標記可以檢測目標基因的有無,無法判斷當代植株的純合或雜合基因型,需要在下一代對基因型或者表型進行群體驗證[4-5,11-12]。以前的檢測方法所需時間較長,對試驗技術條件要求較高,且受儀器、設備和電力以及空間等諸多因素的限制,不利于快速檢測以及在基層實驗室的推廣應用[13-16]。在篩選含多個Bt抗性基因的純合后代時,利用以前的方法進行鑒定效率較低,費時費力。根據轉基因插入位點附近的序列設計多重PCR 標記,可以有效鑒別基因型的純合和雜合[5,17-19],特別是在多價抗性純合后代的篩選中,能提高效率,加快育種進程。
轉Bt基因水稻TT51-1(轉cry1Ab/cry1Ac融合基因),T2A-1(轉cry2A基因)和T1C-19(轉cry1C基因),是培育抗螟蟲水稻和多價抗蟲水稻的優良親本。本研究根據TT51-1,T2A-1 和T1C-19 中外源DNA 插入位點兩側基因組序列分別設計引物L 和R,根據插入序列設計引物I,通過L+R+I 的引物組合檢測結果,不僅可以在單個抗性基因材料中同時鑒定轉基因插入事件的純合、雜合和陰性3 種基因型,還可以在雙價和三價抗蟲材料中,同時鑒定出各個抗性基因的基因型,以期為利用Bt基因進行抗螟蟲水稻分子育種提供一個高效的基因鑒定技術。
1 材料與方法
1.1 材料
轉Bt基因純合系TT51-1(cry1Ab/1Ac)、T2A-1(cry2A)、T1C-19(cry1C)由華中農業大學作物遺傳改良國家重點實驗室/國家植物基因研究中心(武漢)提供。非轉基因水稻9311、R988、超泰B 由本實驗室保存。
分別用9311 與TT51-1,R988 與T2A-1,超泰B 與T1C-19 雜交,以獲得的F1自交得F2用于篩選單價Bt轉基因插入事件的不同基因型植株(圖1-A);同時將三個F1彼此間兩兩雜交,在雜交F2后代中篩選雙價Bt轉基因插入事件的不同基因型(圖1-B);在9311/TT51//R988/T2A-1 的F2后代中篩選出雙價純合TT51-1(+/+)T2A-1(+/+)基因型的水稻單株,與T1C-19(T1C-19(+/+))雜交,在雜交F2后代中篩選三價Bt轉基因插入事件的不同基因型(圖1-C)。
1.2 方法
1.2.1 DNA 提取方法
1.2.1.1 堿裂解法 剪下大約1-2 cm 長的水稻葉片放于2 mL 圓底離心管中,并在其中加入鋼珠和40 μL 0.25 mol/L NaOH 溶液,放在QIAGEN Tissuelyser樣品打樣機中磨碎勻漿,再加入160 μL 0.05 mol/L(pH8.0)Tris-HCl,振蕩混合搖勻后12 000 r/min 離心,取上清,得到DNA。

圖1 用于檢測的不同基因型水稻材料
1.2.1.2 CTAB 法 取約100 mg 新鮮的水稻嫩葉組織放入2 mL EP 管,在液氮冷凍條件下將組織研磨成粉末狀。再加入600 μL 2×CTAB 提取液(2×CTAB提取液使用之前加入0.2%的巰基乙醇),顛倒混勻之后放入水浴鍋中,65℃水浴30-60 min,期間每10 min 顛倒混勻一次。取出EP 管,待其冷卻后加入等體積的600 μL 的24∶1 的氯仿/異戊醇混合液,劇烈振蕩,使之充分混勻,12 000 r/min 室溫離心10 min。然后取上清液約400 μL,加入1/10 體積的3 mol/L NaAC 溶液40 μL,加入2 倍體積的無水乙醇880 μL,充分混勻后于-20℃冰箱中靜置30 min,12 000 r/min,4℃離心10 min 棄上清,再次12 000 r/min,4℃離心1 min,用100 μL 的移液槍吸去管底的多余液體。加入200 μL 75%乙醇,溫和震蕩EP 管,將沉淀彈起,清洗沉淀,8 000 r/min,4℃離心2 min后棄去上清,重復上一清洗步驟。棄去上清,再次離心,用100 μL 的移液槍吸去管底的多余液體,風干沉淀6 min,加入50 μL 1×TE 溶液,用槍反復吹打,確保沉淀溶解完全,得到DNA。
1.2.2 引物設計方法 由NCBI 數據庫中查詢得到TT51-1(Genebank:EU880444.1)的 外 源 片 段插入序列及插入位點兩側水稻基因組序列,T2A-1(GenBank:HQ161063.1)的部分外源片段序列及插入位點左側水稻基因組序列,T1C-19(GenBank:HQ161062.1)的部分外源片段序列及插入位點左側水稻基因組序列。根據TT51-1 插入序列及兩側基因組序列設計L+R 及I+R 組合用于分別檢測基因組片段和插入事件。根據T2A-1 和T1C-19 插入序列及左側基因組序列設計L+I 引物組合檢測插入位點,在插入位點右側選取1 kb 左右基因組片段設計R 引物,用L+R 組合檢測基因組片段(圖2)。所有引物利用Premier5 軟件設計,引物序列及擴增產物信息見表1,由武漢金開瑞生物工程有限公司合成。
1.2.3 二引物PCR 擴增產物測序方法 PCR 產物經1%的瓊脂糖凝膠電泳分離后,回收包含目標片段的膠塊,用南京諾唯贊生物科技有限公司生產的DNA膠回收試劑盒回收目標片段DNA,送北京奧科鼎盛生物科技有限公司進行測序。
1.2.4 不同PCR 擴增體系方法
1.2.4.1 三引物PCR 擴增體系 因為所設計的引物退火溫度均在52-57℃之間,在此區間做梯度PCR測試發現溫度影響較小,最終選用56℃作為體系的退火溫度。
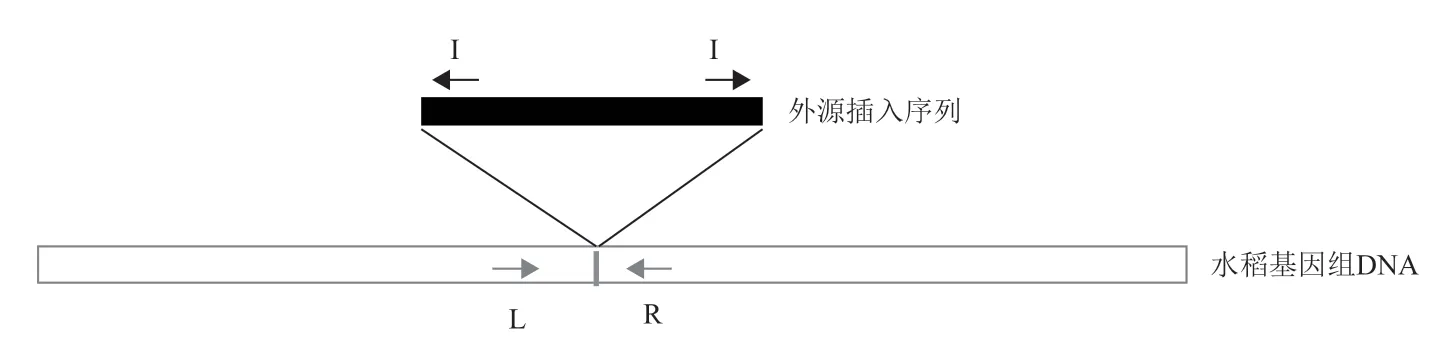
圖2 三引物PCR 檢測轉基因插入事件原理示意圖

表1 實驗所用引物及產物序列
考慮到三引物多重PCR 擴增體系中存在1 個左側引物L、1 個右側引物R 和1 個插入序列I,引物相互之間的競爭對擴增效果會有影響。所以,設計了幾個不同引物使用量的PCR 反應體系,最終選用最為合適的PCR 擴增體系即2AL1∶2AR1∶2AI1按 照0.5∶0.15∶0.35 的 比 例 進 行 混 合 擴 增,1AR1∶1AL1∶1AI1 或1AR1∶1AL1∶1AI2 和1CL1∶1CR1∶1CI1 或1CL2∶1CR2∶1CI2 按 照0.5∶0.15∶0.35 的比例進行混合擴增,分別能得到兩條大小有明顯區別且明亮的條帶。能區分轉基因雜合體、轉基因純合體和非轉基因材料,在雜合體中擴增出清晰的片段,片段大小分別與純合體和陰性材料中的片段大小一致,表現在雜合體兩條片段的擴增量幾乎一致。
轉基因插入位點側翼引物L+R 用于擴增基因組序列,L/R+插入序列特異引物I 用于檢測特異轉基因事件。利用L+R+I 的三引物組合,在轉基因純系材料TT51-1、T2A-1、T1C-19 中,僅能擴出特異轉基因事件條帶。在轉基因陰性材料9311、R988、超泰B 中,僅能擴出基因組序列條帶。在轉基因雜合材料9311/TT51-1、R988/T2A-1、超泰B/T1C-19 中,能擴出兩條預期大小條帶。
PCR 反應體系為30 μL,其中包含2×mix 15 μL(諾唯贊green mix),DNA 模板2 μL 和合適量的三引物L、R 和I(引物濃度為10 μmol/L,引物使用量根據優化結果確定)。PCR 反應程序為95℃反應5 min,然后進入三溫度循環,每個循環包括 94℃反應30 s,56℃反應45 s,72℃反應1 min,共30 個循環,最后72℃反應5 min。
1.2.4.2 六引物PCR 擴增體系 為了測試不同Bt基因的三引物體系能否同時使用以檢測雙價和三價轉基因水稻,將9311/TT51-1、R988/T2A-1 和超泰B/TIC-19 兩兩雜交,在雜交后代中利用3 種三引物體系檢測篩選出不同基因型的雙價水稻單株,以這些后代DNA 作為模板,分別利用不同引物組合進行PCR 擴增。
設計了幾個不同引物使用量的PCR 反應體系,最終選用最為合適的PCR 擴增體系:
檢 測TT51-1和T2A-1的6 條 引 物,1AR1∶1AL1∶1AI1∶2AL1∶2AR1∶2AI1 按 照0.4∶0.1∶0.5∶0.4∶0.1∶0.5 的比例進行混合擴增;檢測TT51-1和T1C-19的6 條 引 物,1AR1∶1AL1∶1AI1∶1CL1∶1CR1∶1CI1 按 照0.5∶0.05∶0.45∶0.5∶0.05∶0.45 的比例進行混合擴增;檢測T2A-1和T1C-19的6 條引物,2AL1∶2AR1∶2AI1∶1CL1∶1CR1∶1CI1 按 照0.4∶0.15∶0.45∶0.4∶0.15∶0.45的比例進行混合擴增。
PCR 反應體系為30 μL,其中包含2×mix 15 μL(諾唯贊green mix),DNA 模板2 μL 和合適量的6條引物(引物濃度為10 μmol/L,引物使用量根據優化結果確定)。PCR 反應程序為95℃反應5 min,然后進入三溫度循環,每個循環包括 94℃反應30 s,56℃反應45 s,72℃反應1 min,共30 個循環,最后72℃反應5 min。
1.2.4.3 九引物PCR 擴增體系 進一步希望利用3 組引物組合同時檢測3 個Bt基因的基因型。將9311/TT51-1 x R988/T2A-1 F2 后代中基因型為雙價純合的單株與T1C-19 雜交,獲得基因型為TT51-1(+/-)T2A-1(+/-)T1C-19(+/-)水稻材料,用于檢測9 條引物組合的擴增效果。在多引物PCR 體系中,隨著引物數量的增減,多重引物之間存在的引物間的相互干擾的難度加劇。因此,針對9 條引物組合,設計了不同引物使用量的27 個PCR 反應體系(表2)。
在三引物的基礎,把各自適合的引物體系混合在一起后進行PCR 擴增,發現可以出現6 條大小不同的目的條帶,但有的太微弱。故考慮調整引物濃度配比,優化后得到最佳引物配比1AR1∶1AL1∶1AI1∶2AL1∶2AR1∶2AI1∶1CL1∶1CR1∶1CI1 為0.4∶0.15∶0.45∶0.4∶0.1∶0.5∶0.4∶0.15∶0.45,PCR 反應體系為30 μL,其中包含2×mix 15 μL(諾唯贊green mix),DNA 模板2 μL 和最適量的9 條引物(引物濃度為10 μmol/L,引物使用量根據優化結果確定)。

表2 三價轉基因材料PCR 不同9 條引物比例擴增體系
PCR 反應程序為95℃反應5 min,然后進入三溫度循環,每個循環包括 94℃反應30 s,56℃反應45 s,72℃反應1 min,共30 個循環,最后72℃反應5 min。
2 結果
2.1 三引物PCR體系對轉基因水稻材料擴增的特異性檢測
以不同類型水稻DNA 為模板,分別利用不同引物組合進行PCR 擴增,均能擴增出預期大小的目的條帶(圖3)。
對引物L+R、L/R+I 和L+R+I 在不同水稻材料中擴增出的片段的測序結果顯示陰性條帶與預期有所差別(表3),原因是我們以NCBI 數據庫中TT51-1、T2A-1、T1C-19 三個轉基因事件的部分插入序列和日本晴的基因組為參考序列設計引物,但實際PCR 擴增檢測的是其他水稻品種,因此陰性條帶存在些許差異;陽性條帶與預期結果完全一致,均能擴增出目標片段。
2.2 不同轉基因水稻材料擴增及條件優化
2.2.1 雙價轉基因水稻材料擴增及條件優化 由圖4 可以看出,不同的6 條引物組合能夠在雙價轉基因材料中擴增出預期大小條帶。
2.2.2 三價轉基因水稻材料擴增及條件優化 由圖5-A 可見,在三價雜合轉基因水稻(TT51-1(+/-)T2A-1(+/-)T1C-19(+/-))中,體系24 的擴增效果最好,6 條擴增條帶均清晰可見。
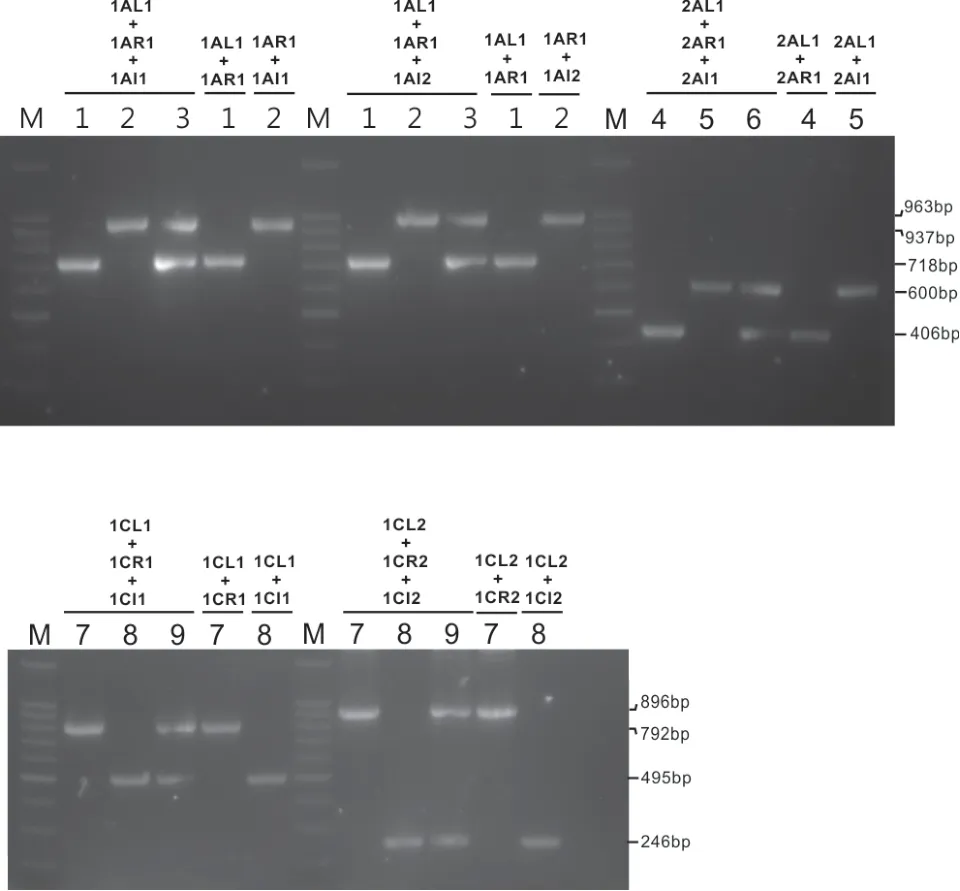
圖3 三引物PCR 體系在水稻材料中的擴增

預期大小/bp 實際大小/bp 9311 716 718 TT51-1 937 937 9311/TT51-1 716 + 937 718 + 937 2AR1+2AL1+2AI1預期大小/bp 實際大小/bp R988 406 434 T2A-1 600 600 R988/T2A-1 406 + 600 434 + 600 1CR1+1CL1+1CI1預期大小/bp 實際大小/bp超泰B 790 792 T1C-19 495 495超泰B/T1C-19 790 + 495 792 + 495
在9311/TT51//R988/T2A-1(TT51-1(+/+)T2A-1(+/+))x T1C-19(T1C-19(+/+))的F2代植株中,利用多引物檢測體系分別篩選含有單價、雙價、三價不同Bt基因型的水稻單株,用于檢測9 條引物檢測體系的特異性。利用體系24 的引物配比,對不同基因型的轉基因材料進行了檢測,結果(圖5-B)表明9 條引物PCR 體系可以準確區分3 種Bt基因的不同基因型。
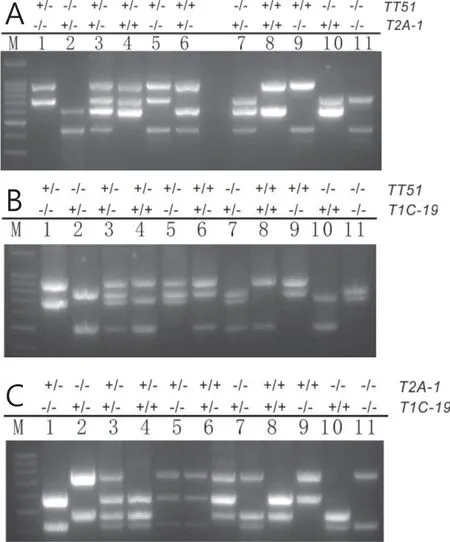
圖4 6 條引物PCR 體系檢測雙價轉基因水稻
同時也檢測了該PCR 體系對不同提取方法所獲得的DNA 模以及對不同商用PCR 擴增試劑的通用性(圖5-C),發現對于堿裂解法粗提的DNA 和CTAB 提取的DNA 作為模板進行擴增效果沒有明顯差別,對不同的商用試劑也有很好的兼容性。
2.3 單價或多價轉基因抗蟲水稻材料的分子檢測
根據雜交親本的基因型狀況不同,利用該檢測方法能夠在F2或者F3代中快速鑒定出單價、雙價和三價Bt轉基因水稻的轉基因純合單株。隨機選取了基因型為TT51-1(+/-)T2A-1(+/-)T1C-19(+/-)自交后代里的288 株,提取DNA 后做9 條引物PCR檢測結果見表4。
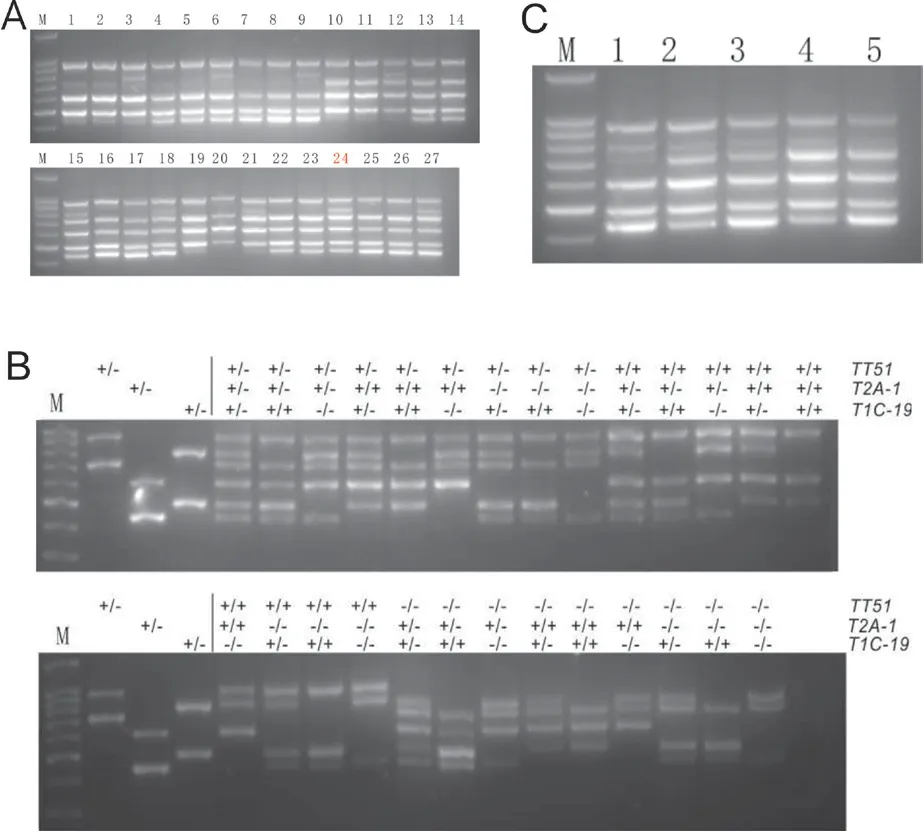
圖5 9 條引物PCR 體系檢測3 價轉基因水稻
3 討論
利用Bt基因能夠增強轉基因植物對螟蟲的抗性,已經在國內外多年的研究和商業化種植得到充分證明。但同時也存在長時間大規模利用單一Bt抗性基因后,使害蟲產生抗性的風險。選育帶有多個不同Bt基因的多價轉基因作物,是降低這一風險的有效手段[5-7]。然而抗蟲性在形狀上通常表現為顯性或者半顯性,因此很難從表型鑒定上來判斷多個Bt基因的導入以及純合/雜合情況。利用分子標記輔助選擇手段,可以在選育后代中檢測多個Bt基因的導入情況,對于這種多個功能類似外源基因導入的選育,具有更明顯的優勢,能夠加快選育進程。常用的Bt基因分子標記,多是根據插入片段設計的特異顯性標記,而無法判斷當代植株的純合或雜合情況[20]。
利用轉基因插入位點附近的序列設計三引物的多重PCR,可以在一個反應中鑒定Bt基因的純合、雜合和轉基因陰性,能大大提高檢測效率[5]。因此利用多重PCR 的方法,理論上也可以同時檢測多價Bt轉基因后代的基因型。多重PCR 可以在 PCR 反應中同時加入多對引物,同時擴增多個目的基因,利用一次 PCR 反應,就能同時檢測多個靶標基因,與傳統 PCR 方法相比,具有低耗時、高通量、節省樣品的優點[21],但同時在引物設計、反應條件優化上也存在著不小的技術難度。在本研究中,我們針對已經獲得轉基因安全證書的轉基因水稻TT51-1(轉cry1Ab/cry1Ac融合基因)、以及在轉基因育種研究中被廣泛使用的供體材料T2A-1(轉cry2A基因)和T1C-19(轉cry1C基因),設計了一系列多重引物,用于在水稻選育材料中檢測單價、二價、三價轉Bt基因的基因型,以便盡快確認Bt基因的純合或者雜合狀態,解決抗蟲表型鑒定無法區分單價和多價的問題,能夠加快抗蟲轉基因水稻的選育進程。
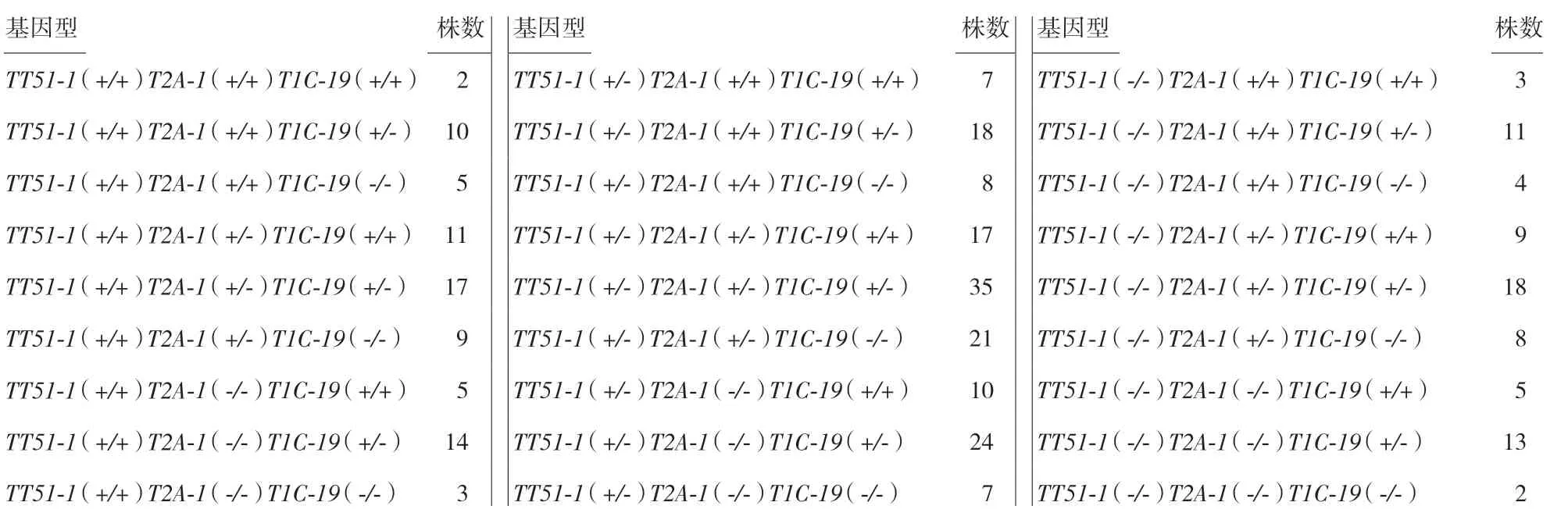
表4 在TT51-1(+/-)T2A-1(+/-)T1C-19(+/-)自交后代中隨機選取288 個水稻樣品,用9 條引物檢測法進行快速鑒別后代基因型的結果統計
多重PCR 的實驗設計難度隨著擴增片段的增加而遞增,要避免引物間的相互結合,引物與模板上目標片段以外的非特異性結合,還要注意不同擴增片段間的擴增效率差異引起的資源競爭以及不同擴增片段的檢測區分[21]。本研究中,我們針對3 個轉化事件合理設計了多重PCR 引物,最多可以同時利用9 條引物檢測3 個Bt基因的轉基因情況。本研究設計的9 條引物PCR 擴增體系產物條帶清晰,條帶大小區分明顯,在普通瓊脂糖凝膠上即可有效區分,不需要借助熒光探針或者聚丙烯酰胺凝膠電泳等較昂貴或較費時的檢測手段。
在聚合多價Bt基因的檢測過程中,會涉及到大量的DNA 提取,按照傳統的CTAB 提取方法費時費力。利用NaOH 和Tris-HCl 能快速提取水稻DNA 用于PCR 檢測,但提取的DNA 質量卻不如傳統CTAB提取方法。在不同PCR 反應體系中的測試結果表明,用常用的國產或者進口PCR 反應試劑均能獲得較好的擴增結果。對DNA 模板的測試結果也表明,不論是經過裂解、抽提、純化的基因組DNA,還是粗提未經純化的基因組DNA,均能得到一致的檢測結果。這些測試說明本次設計的多重PCR 體系,能夠快速、有效、經濟地對大量育種后代進行Bt基因型的檢測,以配合后續的農藝性狀評估,綜合篩選出合適的選育后代。表3 檢測結果只需要一個工作日時間便可獨立完成,從用堿裂解法粗劣提取DNA,到PCR 擴增,跑膠電泳,最終統計結果,體現了本方法的高效性。本實驗結果證明該檢測方法的可靠性,且在不同實驗試劑中具有良好的兼容性。
利用Bt基因進行抗螟蟲水稻育種是防治水稻螟蟲的有效途徑,多價Bt轉基因水稻的選育是抗蟲轉基因水稻品種改良的一個重要方向,多引物多重PCR 方法的建立為利用多價Bt基因抗螟蟲水稻分子育種提供了一個簡便有效的基因檢測技術,將有助于提高對Bt基因的檢測效率,加快抗螟蟲水稻的育種進程。
4 結論
本研究針對多價轉Bt抗蟲水稻設計多重引物及反應體系,可在瓊脂糖膠上快速區分代表TT51-1、T2A-1、T1C-19 事件插入的陽性、陰性條帶,用于對轉Bt基因插入事件水稻的純合、雜合、陰性進行檢測,且對不同的PCR 試劑及不同提取方法的DNA模板都有很好的兼容性。
猜你喜歡
青少年科技博覽(中學版)(2022年6期)2022-12-27 19:44:27
軍事文摘(2021年22期)2021-11-26 00:43:51
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
文苑(2020年6期)2020-06-22 08:41:52
文苑(2019年22期)2019-12-07 05:29:00
海峽科技與產業(2016年3期)2016-05-17 04:32:12
新高考·高一物理(2014年1期)2014-09-18 01:26:07
