HPLC測定不同地區桑椹飲片中七種成分含量△
2019-12-05 06:15:18常艷旭王濤李晉何俊金華馬琳楊雪晶
中國現代中藥 2019年10期
常艷旭,王濤,李晉,何俊,金華,馬琳,楊雪晶,3*
1.天津中醫藥大學/天津市現代中藥重點實驗室,天津 300193;2.天津中醫藥大學 中藥學院,天津 300193;3.哈爾濱商業大學 藥學院,黑龍江 哈爾濱 150076
桑椹為桑科植物桑MorusalbaL.的干燥果穗。現代研究表明,桑椹具有抗氧化、延緩衰老、抗疲勞、增強免疫力、降血脂、降血糖等藥理作用[1]。生物堿、黃酮類和有機酸為桑椹的主要化學成分,如蘆丁有祛脂的效果、具有抗氧化能力[2],綠原酸具有抑菌、抗炎、調節脂質代謝等功效[3-4]。
2015版《中華人民共和國藥典》尚未規定桑椹的含量測定項中指標成分,僅對其浸出物、水分、總灰分進行檢查[5]。本研究選擇了桑椹飲片中7種成分(新綠原酸、綠原酸、隱綠原酸、蘆丁、異槲皮苷、紫云英苷和槲皮素)為指標性成分,采用高效液相色譜法(HPLC),建立桑椹飲片中7種成分同時測定方法,并對國內不同地區、不同批次的桑椹飲片進行測定,擬為桑椹飲片質量控制指標選擇和質量評價提供參考,為桑椹藥用價值進一步開發利用提供質量評價手段。
1 材料與方法
1.1 儀器
粉碎機(吉首市中誠制藥機械廠);離心機(Sigma);天平(德國Sartorius公司,BP121S);Mill-QⅡ型超純水器(Millipore公司);XW-80A渦旋混合器(上海滬西分析儀器廠);SHB-111循環水式多用真空泵(西安太康生物科技有限公司);Agilent 1260型高效液相色譜儀;SB-1000YDTD超聲清洗槽(寧波新芝生物科技股份有限公司)。
1.2 試藥
紫云英苷對照品(批號:MUST-13092001)、綠原酸對照品(批號:MUST-14091701)、隱綠原酸對照品(批號:MUST-13013002)、蘆丁對照品(批號:MUST-11040302)、槲皮素對照品(批號:MUST-13072505)、異槲皮苷對照品(批號:A0439)和新綠原酸對照品(批號:12031401)均購自成都曼思特生物科技有限公司,其純度均>98%。
桑椹飲片購自國內不同省市的藥店,經天津中醫藥大學馬琳教授鑒定為桑科植物桑MorusalbaL.的干燥果穗,樣品保存于天津中醫藥大學中醫藥研究院。桑椹樣品粉碎,過60目篩,備用。
1.3 方法
1.3.1 色譜條件 江蘇漢邦科技有限公司ODS-C18(250 mm×4.6 mm,5 μm);以水(含0.2%甲酸)為流動相A,乙腈為流動相B,梯度洗脫程序為0~36 min,7.6%~7.6%B;40~45 min,12%~13%B;55~70 min,17%~28%B;72~74 min,31%~32%B;76~96 min,38.2%~7.6%B;平衡時間為5 min;柱溫為35 ℃;流速為1 mL·min-1;進樣量為15 μL;吸收波長360 nm。
1.3.2 樣品的制備 精密稱取桑椹粉末0.5 g,置于10 mL具塞錐形瓶中,加入80%甲醇10 mL,稱質量,超聲提取40 min,靜置放冷,補足質量,14 000 r·min-1離心10 min后,取上清液,過0.22 μm微孔濾膜,即為樣品溶液。
1.3.3 標準曲線及線性范圍 將對照品儲備液配制成含有新綠原酸(250 μg·mL-1)、綠原酸(1.4 mg·mL-1)、隱綠原酸(1 mg·mL-1)、蘆丁(800 μg·mL-1)、異槲皮苷(150 μg·mL-1)、紫云英苷(100 μg·mL-1)、槲皮素(100 μg·mL-1)的混合對照品儲備液。精密移取該混合對照品儲備液,用甲醇稀釋成一系列濃度,記錄各濃度下的峰面積,并制作標準曲線。
1.3.4 精密度、穩定性、重復性試驗 精密度分為日間精密度和日內精密度。日內精密度為同一份樣品溶液在1 d內連續進樣6次,日間精密度為連續3 d內每天連續進樣6次。穩定性試驗為一份樣品溶液分別在0、2、4、6、8、12、24 h內進樣。重復性試驗為平行制備6份桑椹樣品溶液,連續進樣。
1.3.5 加樣回收率 精密稱定已知含量的桑椹藥材粉末0.250 g(平行操作6份),分別按所含7種成分量的50%加入對照品,按照樣品處理方法進行處理,測定加樣后7種成分的含量,計算加樣回收率。
2 結果
2.1 高效液相色譜法方法學驗證
2.1.1 標準曲線的繪制 以對照品質量濃度為橫坐標(μg·mL-1),峰面積為縱坐標制作標準曲線,并得出回歸方程、相關系數(見表1)。分別以信噪比(S∶N)3∶1和10∶1作為檢測限(LOD)和最低定量限(LOQ)。從表1中可以得知,7個化合物在線性范圍內相關系數均大于0.998 0,表明7個化合物在各自的線性范圍內線性關系良好。
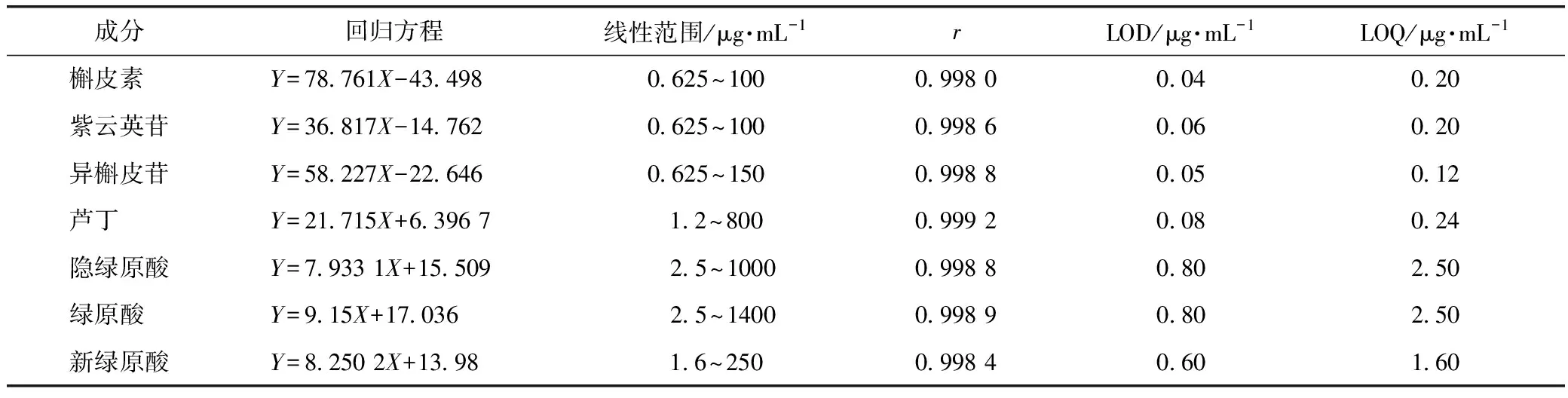
表1 7種成分標準曲線、線性范圍、LOD和LOQ
2.1.2 精密度、穩定性、重復性試驗 桑椹中各成分的精密度、重復性和穩定性結果見表2。結果發現7種成分日內、日間精密度RSD<4.9%,表明該方法精密度良好;重復性RSD<2.4%,表明該方法重復性良好;穩定性RSD<3.0%,表明該方法穩定性良好。
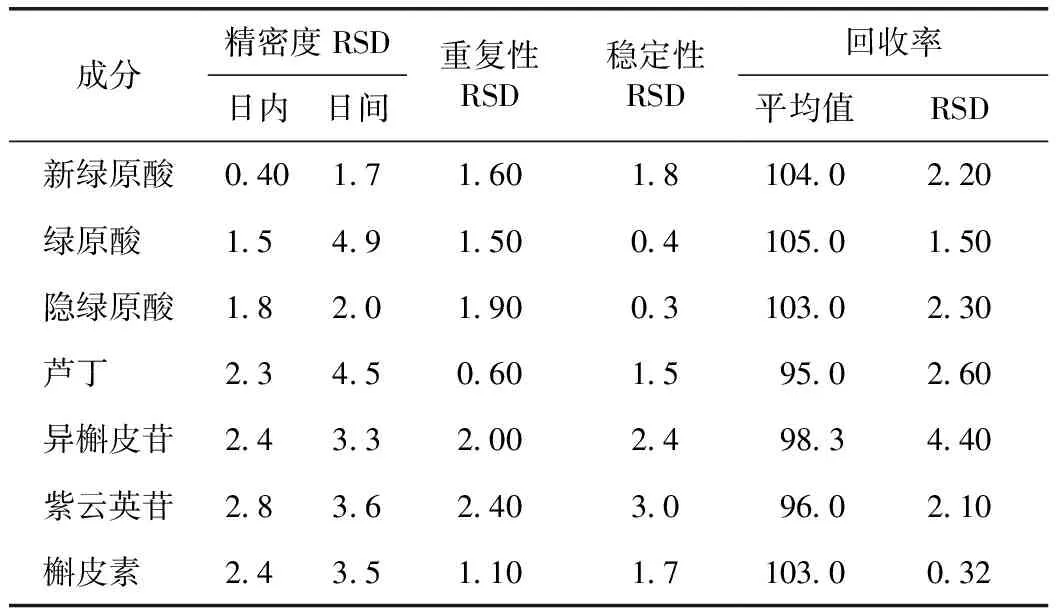
表2 7種成分精密度、重復性、穩定性和回收率結果 %
2.1.3 加樣回收率 由表2結果可知,7種成分的平均回收率均在95.0%~105%,且RSD<4.4%,表明該方法對桑椹中7種成分的含量測定的準確度較高。
2.2 桑椹飲片中7種化學成分含量
桑椹飲片樣品中7種成分以及對照品溶液的色譜圖見圖1。由表3可知,桑葚飲片中各成分質量分數:新綠原酸10.96~478.82 μg·g-1;綠原酸5.82~2 619.6 μg·g-1;隱綠原酸9.56~1 657.58 μg·g-1;蘆丁26.8~1 307.58 μg·g-1;異槲皮苷7.78~253.48 μg·g-1;紫云英苷8.02~62.72 μg·g-1;槲皮素11.04~68.64 μg·g-1。
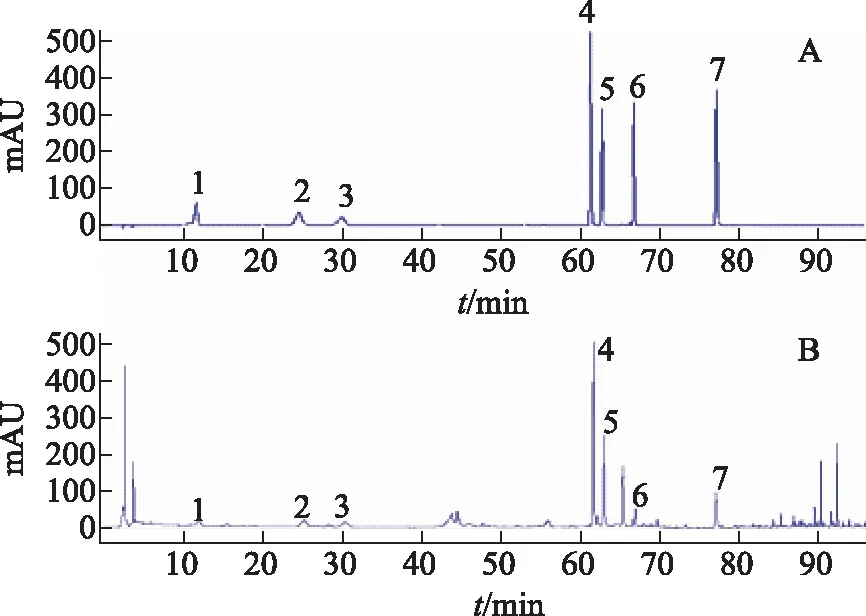
注:A.混合對照品溶液;B.桑椹飲片樣品溶液;1.新綠原酸;2.綠原酸;3.隱綠原酸;4.蘆丁;5.異槲皮苷;6.紫云英苷;7.槲皮素。圖1 混合對照品和桑椹飲片的HPLC圖
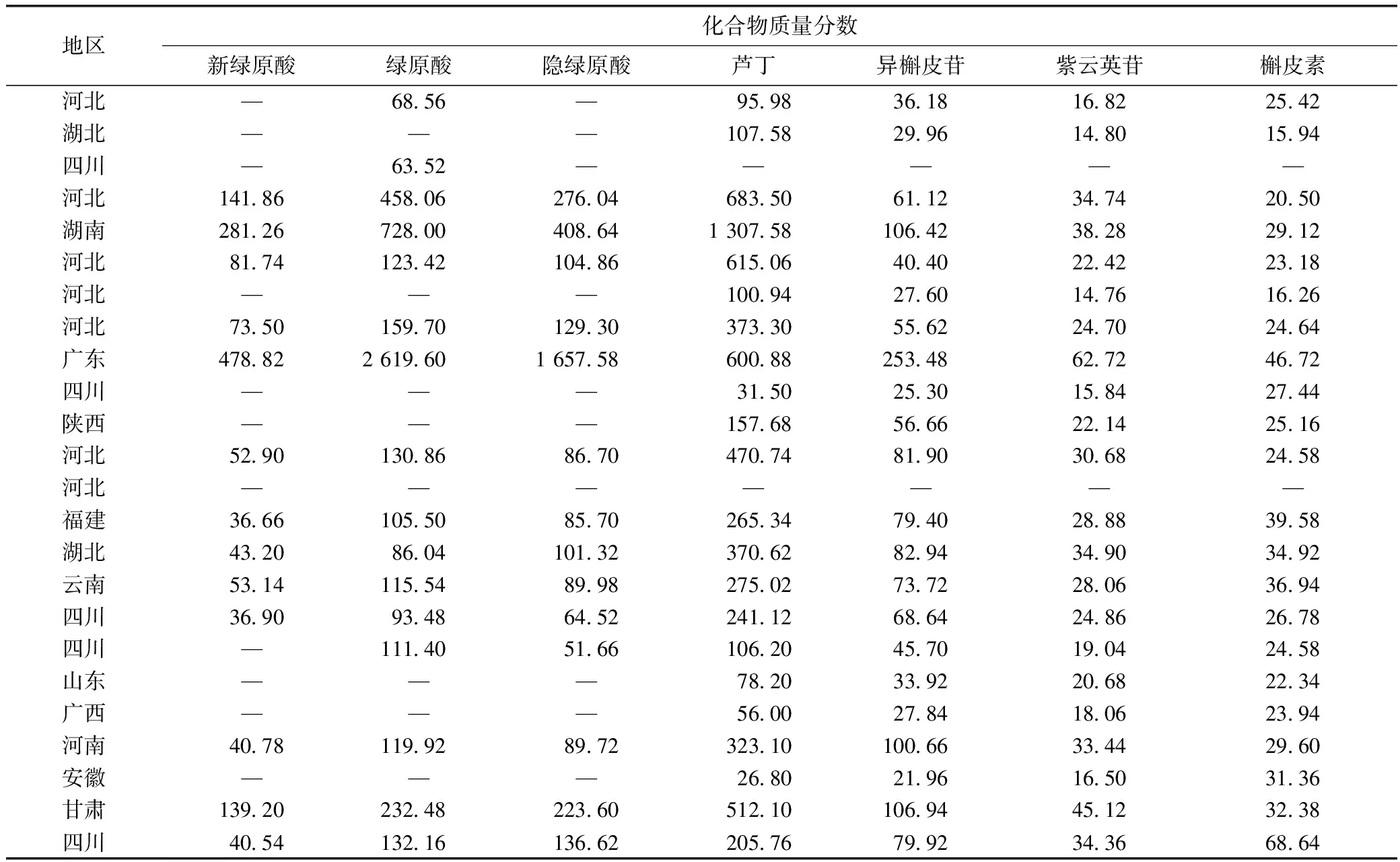
表3 不同地區的桑椹飲片7種化合物含量測定結果(n=3) μg·g-1
注:—為對應化合物的含量在檢測限下或該批次飲片中不含有該種成分。
3 討論
3.1 流動相選擇
在選擇流動相過程中,因為桑椹中化合物的極性范圍分布較廣,且化合物種類繁多,在色譜分離時不易得到理想的峰形,參考相關文獻[6],考察了0.1%甲酸水-乙腈、0.1%甲酸水-0.1%甲酸乙腈、0.1%磷酸水-乙腈、0.1%磷酸水-甲醇、0.1%磷酸水-甲醇乙腈(1∶1∶1)和0.2%甲酸水-乙腈等流動相組成,發現水相為磷酸水的效果要優于甲酸水,峰形較好,且分離度良好,同時整體的色譜分離檢測時間稍短。有機相乙腈的效果好于甲醇-乙腈(1∶1)、甲醇,其中加了甲酸的乙腈的色譜圖基線較不加甲酸的更加平穩,但無明顯影響,因此選擇了0.2%甲酸水-乙腈為流動相。
3.2 提取條件的選擇
采用飲片粉碎后用超聲波提取,該方法具有簡便、安全、有效等特點。在單因素提取條件下,以提取效果為指標,提取時間結果為:40 min>20 min>30 min>10 min;溶劑濃度:60%>80%>100%>40%;料液比:1∶20>1∶15>1∶10>1∶5。運用正交優化試驗法,選取甲醇濃度、提取時間、料液比3個因素,每個因素為3水平,以桑椹中新綠原酸、隱綠原酸、綠原酸、蘆丁、異槲皮苷、紫云英苷和槲皮素7種成分的總含量為指標,選用L9(34)正交設計,對桑椹中7種成分最佳超聲提取條件的優化,發現甲醇濃度是最關鍵的影響因素,其次為料液比和提取時間;確定最佳提取條件為0.50 g桑椹粉末采用10 mL 80%甲醇溶液超聲提取40 min。
4 結論
本研究建立了一種穩定、可靠的桑椹中7種黃酮類成分(新綠原酸、綠原酸、隱綠原酸、蘆丁、異槲皮苷、紫云英苷和槲皮素)含量同時測定的高效液相色譜(HPLC)方法,經方法學驗證,符合分析要求,并對不同地區、不同批次的桑椹飲片中7種成分的含量進行測定,結果表明,蘆丁為平均含量最高成分,而異槲皮苷、紫云英苷、槲皮素平均含量較低,國內不同地區所用桑椹飲片質量存在較大差異,需要對其生產產地、加工方法、質量評價指標進行深入研究,確保其臨床有效性。
