溶液緩沖劑發射光譜法在地質樣品多元素測定中的應用
2019-12-13 02:03:46
分析儀器 2019年6期
關鍵詞:方法
(北京北分瑞利分析儀器(集團)有限責任公司,北京市物質成分分析儀器工程技術研究中心、北京市企業技術中心,北京 100095)
1 引言
電弧發射光譜法由于其可同時、快速測定復雜基體的巖礦中的多種元素,且測定檢出限低,測定范圍較寬,在地質化探領域一直占據不可替代的位置。
北京北分瑞利分析儀器(集團)有限責任公司研制出的AES-8000全譜交直流電弧發射光譜儀是專門為地質、有色等行業研發的直讀光譜儀。地質工作的巨大樣品量不僅需要快速測定的直讀光譜儀,更需要簡單快速的測定方法來減輕工作量。現地質行業主流方法是廊坊物探所的《發射光譜法測定勘察地球化學樣品中銀硼錫鉬鉛》[1],其采用的是固體緩沖劑(后面出現的“固體緩沖劑”均特指此文中的緩沖劑。)。
以溶液作為緩沖劑的方法早已問世[2],該以溶液形式加入到試料電極中測定銀、鉬等。由此可知,加入溶液緩沖劑進行光譜定量測定不是什么難題,但它的簡便、實用卻引人注目。
為提高測量效率,現選用氟化銨溶液作為緩沖劑,省去了最為耗時的稱量和磨樣的前處理時間,大大提高了工作效率,也節約了大量固體緩沖劑的成本,更加環保。且該方法在保證Ag、Sn、B指標變化不大的前提下,增加了Be、Co、Cr、Ni、V等多元素的同步定量測定,滿足了用戶進一步的要求。
由實驗結果可以看出,本方法對Ag、Sn、B、Mo、Pb、Be、Co、Cr、Ni、V元素檢測的準確度較高,且同時適用于水系沉積物和土壤樣品的測定,可以滿足用戶的日常生產需求。
2 實驗部分
2.1 儀器與工作條件
2.1.1儀器及參數
AES-8000全譜交直流電弧發射光譜儀(北京北分瑞利分析儀器(集團)有限責任公司),Ebert-Fastic光學系統分光,CMOS檢測器,光柵刻線密度2400條/mm,焦距600mm,線色散率達到0.64nm/mm,理論光譜分辨率達到0.0031nm(300nm),光欄3.2mm,自動對準、自動控溫水冷電極夾[3]。
上、下電極對光時,上電極距光欄上沿1mm,下電極距光欄下沿3mm。
2.1.2光源
交直流電弧發生器,交流電弧,4.5A預燃,5s后切換為14.5A,保持40s曝光。
2.1.3標準物質系列
合成硅酸鹽光譜分析標準物質(GBW07701~GBW07709),基物(地礦部物化探研究所)。
2.1.4試樣制備
標準物質系列、基物和樣品處理方法相同,裝入下電極中壓實,碾平電極口。先滴加一滴蔗糖溶液以利于緩沖劑滲透,且能有效防止激發時樣品的噴濺。再滴加1滴氟化銨溶液,滴液時保持液珠覆蓋在樣品上,不流出電極頭,待滲透充分之后,100℃烘干備用。
液體緩沖劑取樣器采用一次性塑料滴管,同批樣品用同一滴管,取樣量的差異對實驗結果影響不大。
氟化銨在水中水解出少量氫氟酸能與玻璃反應溶解出少量硼,作為液體緩沖劑會嚴重影響樣品中微量硼的測定,實驗中也證明了這一點,譜圖顯示硼元素譜峰強度巨大。所以氟化銨溶液配制及儲存都應用塑料(如特氟龍)制品,如滴管、移液器、燒杯、容量瓶等。
3 結果與討論
3.1 電極的選擇
做深電極(?4mm×6mm×0.7mm)和普通電極(?3.8mm×4mm×0.6mm)的對比實驗。當采用深電極時,樣品量增加,倍數和檢出限有明顯提高,但是相應的蒸發時間拉長,背景加深,弧焰不穩定,導致重復性和準確度變差。故選用普通電極。
3.2 緩沖劑的選擇
3.2.1溶液緩沖劑對分析元素蒸發的影響
陰離子效應:選用氟化銨液體緩沖劑,利用元素的鹵化反應,使待測元素形成易揮發的鹵化物,縮短蒸發時間[4],分析元素相對的得到分離富集,減弱或消除了基體元素的干擾,使得分析元素與基體元素得到有效分餾;而待測元素具有相似的蒸發行為,蒸發時間相近,則待測元素之間的分餾效應被消除。從而提高了分析元素的靈敏度和準確度。
陽離子效應:電弧激發時,銨鹽大部分揮發,部分被分解,出現高激發電位氮分子相對富集[5],形成了一個穩定的等離子體,這穩定了光源和電弧溫度,并改善了弧焰中蒸發物質的分布,延長了待測元素在弧焰中的停留時間,使分析元素得到相應的富集,從而使其譜線強度增大,降低了分析元素的檢出限。
3.2.2緩沖劑濃度的選擇
選擇10%(1滴、2滴)、15%(1滴、2滴)、20%(1滴)和30%(1滴)進行實驗。對比后發現,10%(2滴)、15%(1滴)和20%(1滴)指標較好,相差并不很明顯。但2滴的滲透時間過長且容易流液,增加了前處理的時間成本和難度,故選擇1滴。20%的緩沖劑滲透后在電極頭表面形成的結晶過多,使激發時弧焰不穩定,重復性較差。15%的緩沖劑對于5元素的倍數和準確度都比20%的緩沖劑要好,且濃度低易于配制和滲透,更環保,故選擇15%的NH4F溶液作為緩沖劑。
3.2.3倍數
算出合成硅酸鹽光譜分析標準物質GSES I-7(ST-7)絕對強度GSES I- 1(ST-1)絕對強度的倍數,可看出梯度大小。如表1所示。
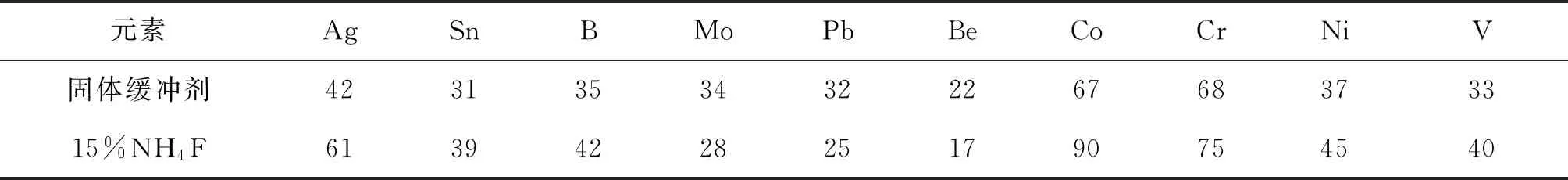
表1 固體緩沖劑與氟化銨液體緩沖劑倍數對比(ST-7/ST-1)
氟化銨液體緩沖劑的Ag、Sn、B元素倍數相對于固體緩沖劑均有提高;其他元素,除Mo、Pb、Be外,倍數也均有所提高。與固體緩沖劑相比,液體緩沖劑的裝樣量增加了一倍左右,所以相應的待測元素絕對強度增加明顯,背景降低,靈敏度提高。氟化銨緩沖劑有利于多數元素的蒸發,但對個別元素蒸發效果不是很理想。
3.3 蒸發曲線和積分時間的選擇
為了圖表清晰,現只顯示主要元素(Ag、Sn、B)及基體元素(Fe、Mg、Ca)的蒸發曲線,如圖1所示。

圖1 主要元素(Ag、Sn、B)及基體元素(Fe、Mg、Ca)的蒸發曲線
根據蒸發曲線判斷,大部分分析元素在36s前已經基本蒸發出來,而Cr和V蒸發高峰較晚,Be則較快(25s),所以采取分段積分的方法,分別對不同元素截取合適的曝光時間,進行積分。積分時間過長會加深背景,過短則會使重復性變差。所以積分時間的選擇很重要。
從圖中也可以看出,氟化銨液體緩沖劑對于待測元素與基體元素的分餾效應。
根據蒸發曲線確定各個元素的積分時間,如表2所示。

表2 各元素的積分時間
對于基體組成復雜的地質樣品,一般采用內標法進行光譜定量分析。利用內標的光強來控制分析元素譜線強度的變動,從而消除或減小由于光源波動、元素蒸發行為、譜線激發過程等各種干擾因素對譜線強度的影響[6]。由于液體緩沖劑不含有內標元素,故不能利用該方法進行重復性校正,重復性會受到一定的影響。
3.4 工作曲線
用合成硅酸鹽光譜分析標準物質(GSES I-1~ GSES I-9 即ST-1~ST-9)建立標準曲線。
3.5 方法檢出限
按照本方法,對合成硅酸鹽光譜分析標準物質(GSES I)即基物滴加緩沖劑處理后,平行測定12次,即相當于進行12次樣品空白測定,測定結果以3倍標準偏差計算得到方法檢出限,如表3所示。

表3 氟化銨液體緩沖劑各元素的檢出限 μg/g
從表3結果可以看出,Ag, Sn, B,Mo, Pb, Be, Co, Cr, Ni, V均能滿足DZ/T 0130-2006地質礦產實驗室測試質量管理規范的檢出限要求[7]。
3.6 控樣的校正作用
由于實際樣品的基體比合成硅酸鹽光譜分析標準物質的基體復雜許多,且各不相同,故選用7個國家一級標準物質(水系沉積物、土壤和巖石系列)作為控樣(以三角▲顯示),對標準曲線進行校正。Sn、V校正前后曲線和準確度對比,如圖2、3、4、5和表4所示,可以看出,校正后數據的準確度更好。對于含量均較低的錫,控樣離標線較遠,控樣的校正作用較大,校正前后的準確度相差較大;而對于倍數和含量均較高的釩,控樣和標線基本重合,校正幅度較小,校正前準確度基本在合格范圍內,校正前后數據差別不是很大。
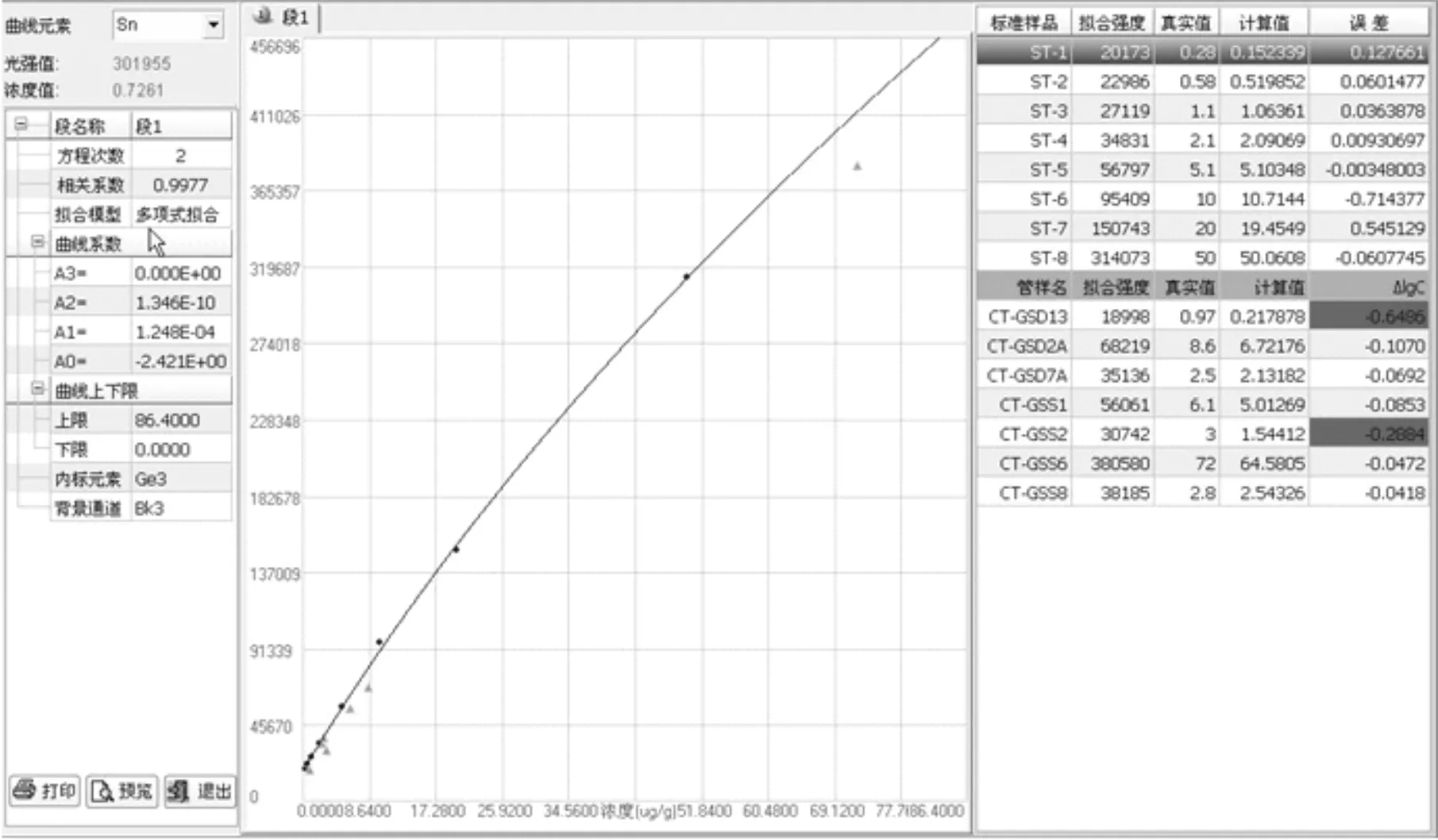
圖2 Sn校正前曲線
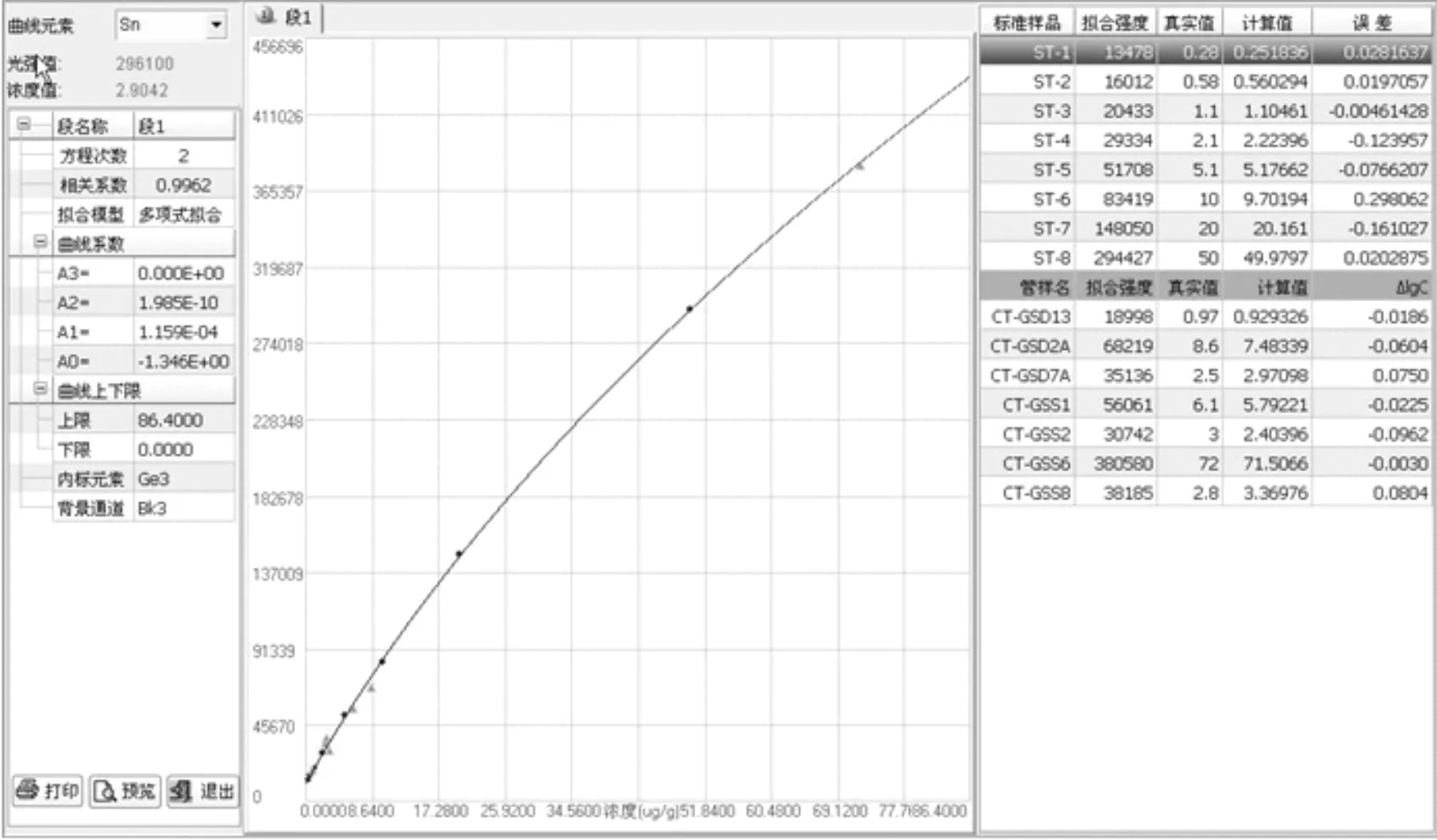
圖3 Sn校正后曲線

圖4 V校正前曲線
圖5 V校正后曲線
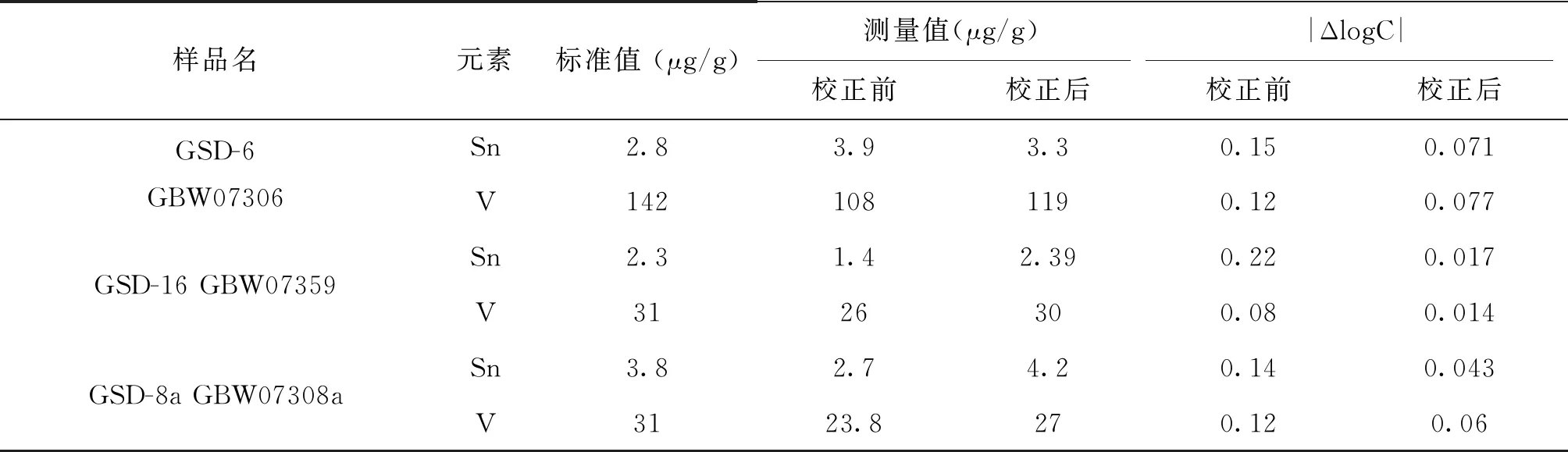
表4 對比校正前、后Sn、V的準確度
3.7 方法重復性和方法準確度
3.7.1公式
(1)
(2)
式中:Ci為每個GBW標準物質12次實測值的平均值;Cs為GBW標準物質的標準值;n為每個GBW標準物質測量參數;Ci為每個GBW標準物質單次實測值。
3.7.2方法重復性
以12個國家一級標準物質(GBW水系沉積物、土壤和巖石)進行重復性實驗,每個樣品進行12次平行測定,相對標準偏差(RSD)結果如表5所示。
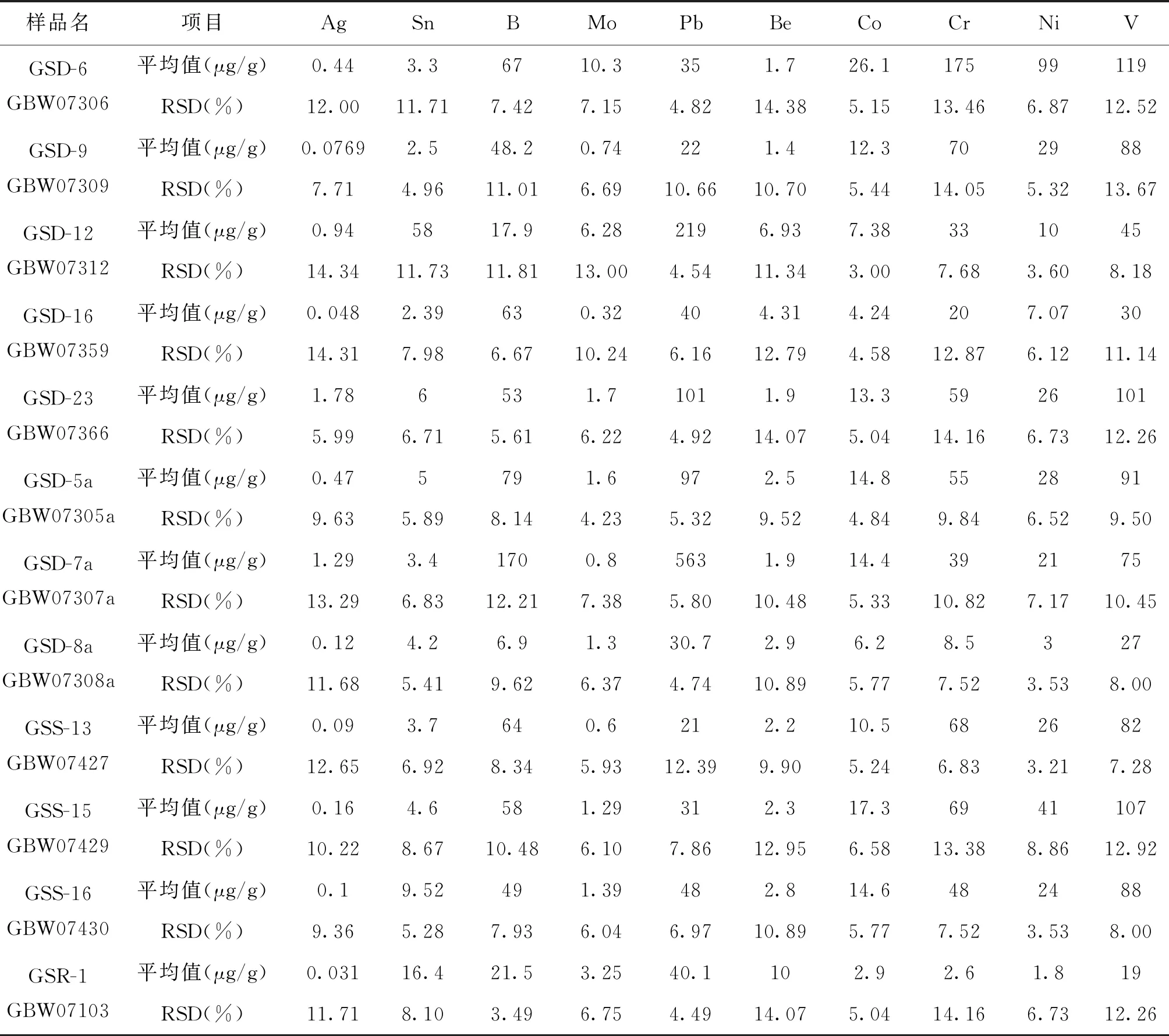
表5 方法的重復性
從表中可以看出,實驗結果的重復性均小于15%,具有較好的重復性,可以滿足地質生產單位一般樣品的重復性要求。
3.7.3方法準確度
測定國家一級標準物質(水系沉積物、土壤)中各元素的含量,結果如表6所示。

表6 方法的準確度
由實驗結果可以看出,本方法準確度較高,且檢測范圍較寬,可以滿足DZ/T 0130-2006地質礦產實驗室測試質量管理規范中1∶50000的要求[6],且本方法同時適用于水系沉淀物、土壤樣品和部分巖石樣品的測定。
4 結論
本方法經過國家標準樣品和用戶野外實際樣品試驗,對于以硅酸鹽為主的巖石、土壤和水系沉積物樣品,測試結果比較可靠。且前處理簡單、快捷,適于地質生產型單位大量化探、找礦樣品的批量測定,省去了大部分的人力和時間成本。
但本方法僅經過小批量樣品檢驗或部分元素經過大批量樣品檢驗,還有必要在生產中多次大批量實踐,以充分證明其可靠性。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
