IgA和IgM雙克隆型淋巴漿細胞淋巴瘤1例報道
2019-12-26 09:25:00郭振興
國際檢驗醫學雜志 2019年24期
鄭 力,郭振興
(清華大學第一附屬醫院血液腫瘤科,北京 100016)
淋巴漿細胞淋巴瘤(LPL)是一種少見的惰性成熟B細胞淋巴瘤,90%~95%的LPL分泌單克隆性IgM,稱為華氏巨球蛋白血癥(WM),僅小部分LPL分泌單克隆性IgA、IgG或不分泌單克隆免疫球蛋白[1]。現首次報告1例同時分泌IgA和IgM雙克隆型LPL,并結合文獻進行復習,以提高對此類疾病的認識水平。
1 臨床資料
患者,男,86歲,主因“乏力7年,加重伴納差3月,發熱1 d”于2018年4月12日入住本院。患者2011年無明顯誘因出現乏力,外院發現重度貧血,腎功能不全(具體不詳),血紅蛋白最低為42 g/L,給予補充造血原料、促紅細胞生成素(EPO)、間斷輸血支持治療后,血紅蛋白維持在50~65 g/L。2018年1月,患者乏力加重,活動耐量下降,伴納差、反酸,無腹痛、腹瀉,無牙齦出血,無黑便、鮮血便,無咯血、嘔血,無血尿。就診當地醫院,查血常規提示:重度貧血(具體不詳),診斷為“腎性貧血”,予EPO(10 000 IU皮下注射QW)治療,同時補充鐵劑、葉酸、維生素B12等治療,效果欠佳。入院前1天,患者無明顯誘因出現發熱,體溫最高為37.7 ℃,無畏寒、寒戰,無咳嗽、咳痰,無盜汗、消瘦,為進一步診治入院。既往有慢性支氣管炎、腎囊腫、慢性腎功能不全病史。2013年患腦出血,無后遺癥。入院查體:體溫 37.7 ℃,心率106次/分,呼吸 18次/分,血壓118/66 mm Hg,神清語利,重度貧血貌,全身淺表淋巴結未及腫大,胸骨中下段無壓痛,雙肺呼吸音低,未聞及干濕羅音,心界不大,心率106次/分,律齊,各瓣膜聽診區未聞及病理性雜音。腹軟,肝脾肋下未觸及,雙下肢無水腫。入院后查血常規:白細胞 2.75×109/L,血紅蛋白36 g/L,平均紅細胞體積115.5 fL,網織紅細胞比例2.12%,血小板142×109/L。腎功能:尿素氮20.40 mmol/L,肌酐 347.0 μmol/L。肝功能及乳酸脫氫酶未見異常。C反應蛋白 5.55 mg/L。血β2微球蛋白 17.84 mg/L,尿β2微球蛋白>88.0 mg/L。24 h尿總蛋白1 019 ng/L,24 h尿清蛋白 73.7 ng/L。貧血組合示鐵蛋白、維生素B12 、葉酸不低。肝炎系列陰性。大便潛血(-)。腹部彩超:右腎囊腫。考慮漿細胞腫瘤,行免疫固定電泳:免疫球蛋白及尿輕鏈測定:IgA 969 mg/dL,IgM 571 mg/dL,IgG 614 mg/dL,IgE<18.2 IU/mL,κ輕鏈1 580 mg/dL,λ輕鏈186 mg/dL。尿蛋白κ輕鏈90.4 mg/dL,λ輕鏈<5 mg/dL;免疫固定電泳中可見M成分IgA-κ;IgM-κ,尿κ輕鏈定量高于參考范圍(圖1)。進一步骨髓細胞形態學結果顯示:骨髓增生明顯-極度活躍;粒系占13.4%;紅系占7.4%;淋巴細胞占76.8%,大部分細胞形態異常:胞體小,染色質不均勻、呈塊狀,胞質量少或極少,邊緣比較整齊。少部分細胞胞漿深染,泡沫感,呈漿細胞樣改變。少部分細胞隱約可見核仁;漿細胞占1.2%,雙核漿細胞可見;全片見巨核細胞89個。考慮:淋巴瘤可能(圖2)。骨髓活檢:骨髓有核細胞增生較活躍,三系造血細胞少量殘存,可見淋巴細胞樣細胞彌漫增生,細胞體積小,胞質豐富,淡染,核中等偏小,類圓或不規則,染色質粗塊狀,核仁不明顯,分裂相少見,部分細胞可見漿樣分化,診斷考慮:非何杰金氏淋巴瘤,成熟小B細胞類淋巴瘤可能性大(圖3)。骨髓單個核細胞流式細胞術檢測:(1)44.37%細胞(占有核細胞)表達ckappa dim、CD19、bcl-2,部分表達CD38、CD20、CD180、CD79b,不表達CD2、CD117、CD56、CD3、CD4、CD8、CD7、clambda、CD229、CD138、CD11c、kappa、lambda、CD5、Ki67、CD10、FMC7、CD103、CD25、CD23、TDT、CD200,為惡性單克隆成熟B細胞,細胞小。(2)1.16%細胞(占有核細胞)表達ckappa、CD38、CD138、CD229dim,部分表達CD19,不表達lambda、CD56,為單克隆漿細胞。考慮為有漿細胞分化的成熟小B細胞淋巴瘤。染色體核型:46,XY[19];另見一個核型為:46,XY,del(2)(q21)[1]。進一步查骨髓MYD88基因突變:檢測到MYD88基因c.794T>C/P.L265P位點突變。結合上述實驗室檢查,患者診斷為:IgA和IgM雙克隆型LPL。因患者高齡,患者及家屬放棄化療及靶向治療,予間斷輸注壓積紅細胞、應用EPO等對癥支持治療。目前隨訪中。
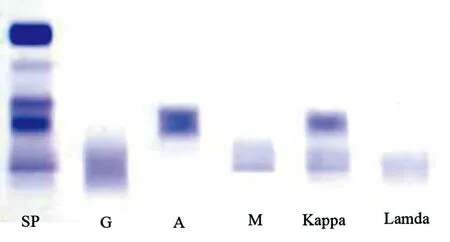
注:血清蛋白電泳及免疫固定電泳中可見M成分IgA-κ;IgM-κ
圖1M蛋白測定

注:大部分淋巴細胞形態異常,少部分呈漿細胞樣改變,雙核漿細胞可見(HE染色)
圖2骨髓細胞形態學

注:可見淋巴細胞樣細胞彌漫增生,部分細胞可見漿樣分化
圖3骨髓活檢
2 討 論
LPL是一種少見的惰性成熟B細胞淋巴瘤,在非何杰金氏淋巴瘤中所占比例<2%。在世界衛生組織(WHO)淋巴與造血組織分類中,LPL定義為由小B淋巴細胞、漿細胞樣淋巴細胞和漿細胞組成的淋巴瘤,通常累及骨髓、淋巴結和脾臟,同時不符合其他任何一種可能伴漿細胞分化的小B細胞淋巴瘤診斷標準[1]。LPL是一種兼具B淋巴細胞、漿細胞特點的少見的特殊類型的非何杰金氏淋巴瘤。由于此病發病率低,臨床表現多樣,故極易誤診。90%~95%的LPL分泌單克隆性IgM,稱為WM。僅小部分LPL分泌單克隆性IgA、IgG成分或不分泌單克隆免疫球蛋白[1]。該患者血清免疫固定電泳中可見M成分IgA-κ及IgM-κ,為雙克隆型,極為罕見。YI等[2]曾報告4例LPL患者同時分泌IgM和IgG異常免疫球蛋白,目前尚無同時分泌IgM和IgA異常免疫球蛋白的LPL病例報道。鄒德慧等[3]對13例非IgM型 LPL和120例WM患者臨床資料進行了回顧性分析總結,CAO等[4]對比了17例非IgM型LPL患者和312例典型WM患者的臨床資料,得出了相同的結論:非IgM型LPL患者和IgM型LPL患者具有相似的臨床和生物學特征。目前,非IgM型LPL的診治參照IgM型LPL(WM)進行[1]。LPL患者診斷時最常見的癥狀是乏力和易疲勞,這主要和貧血相關[5-6]。該患者乏力7年,以重度貧血及腎功能不全為主要臨床表現,貧血考慮為大量小B細胞、漿細胞樣淋巴細胞及漿細胞廣泛的骨髓浸潤所致。該患者尿蛋白κ輕鏈明顯升高,腎功能損傷考慮為過多輕鏈從腎小球濾過后被腎小管重吸收對腎小管的損傷所致。WM表達B細胞相關抗原,例如:CD19(+),CD20(+),CD22(+),CD5(+/-),CD10(-),CD23(-),CD103(-)[1,6]。CD38和CD138常表達于單克隆漿細胞表面[4,7]。該患者CD19(+),CD20(+),CD5(-),CD10(-),CD23(-),CD103(-),為惡性單克隆成熟B細胞,極少部分細胞表達CD38及CD138,考慮為有漿細胞分化的成熟小B細胞淋巴瘤。MYD88-L265P基因突變的檢測對LPL的診斷及治療具有重要意義。90%以上的WM患者存在功能獲得性MYD88-L265P基因突變[1,6-8]。非IgM型LPL MYD88L-265P突變的頻率低于IgM LPL(WM),但也是其標志性分子生物學特征[4,7]。該患者基因MYD88-L265P陽性,進一步支持LPL診斷。
LPL具有低度惡性B細胞淋巴瘤特點,病程較長,經治療可緩解,但不能治愈,自然病程約為5~10年。“觀察和定期復查”是無癥狀LPL患者的標準治療策略[6,9]。由華氏巨球蛋白血癥國際工作組第2次會議(IWWM-2)提出,第8次IWWM確認,WM開始治療的指征為IgM相關并發癥和/或腫瘤侵犯骨髓相關癥狀如全血細胞減少/軀體癥狀,以及髓外大包塊[10]。利妥昔單抗與化療藥物的聯合方案是WM/LPL患者的首選治療方案[1,6,8,10]。Benda-R方案(苯達莫司汀聯合利妥昔單抗)和CHOP-R方案(環磷酰胺、多柔比星、長春新堿及潑尼松聯合利妥昔單抗)有效率相似(95%),但Benda-R方案無進展生存期(PFS)更長,且耐受性更好[6,10]。高黏滯綜合征患者應進行血漿置換[7,10],單藥利妥昔單抗可導致一過性IgM水平升高,故這些患者第一周期化療應避免應用利妥昔單抗,或聯合苯達莫司汀或硼替佐米[6,9]。蛋白酶抑制劑對WM患者有效率很高[6,8-9],國外有文獻報道BDR(硼替佐米+利妥昔單抗+地塞米松)有效率為85%,但46%的患者出現了周圍神經病變[9]。第二代蛋白酶抑制劑卡非佐米的神經毒性風險明顯減低[6,8-10]。BTK抑制劑依魯替尼是FDA唯一批準用于治療WM的藥物,可用于初治及復發患者[6,8-10]。干細胞移植用于年輕、多次復發或原發耐藥的患者[10]。該患者因高齡及經濟因素,家屬放棄化療及靶向治療,采取姑息支持治療,目前已存活7年。
3 小 結
綜上所述,LPL是一種少見的惰性成熟B細胞淋巴瘤,隨著分子遺傳學的進一步研究及新藥的研制,LPL的診療方法將更加完善。研究者首次報道1例IgA和IgM雙克隆型LPL,由于該病極其罕見,今后的臨床實踐中需要進一步積累資料,提高對該病的認識水平。
