NLRC4炎癥小體研究進展①
2019-12-27 06:18:20陳麗嫦于化鵬吳玲玲曾冠盛
中國免疫學雜志 2019年22期
關鍵詞:研究
陳麗嫦 于化鵬 吳玲玲 曾冠盛
(南方醫科大學珠江醫院,廣州 510280)
NLRC4炎癥小體(NLR family,CARD domain containing4,NLRC4)是一種多蛋白復合體,由N-端的含有胱天蛋白酶募集結構域(Caspase recruitment domain,CARD),中間的核苷酸結合結構域(Nucleotide binding oligomerization domain,NOD)和富含亮氨酸的C-端蛋白相互作用結構域(Leucine rich repeats,LRR)構成。在過去的10余年里,研究發現炎癥小體的活化能夠激活半胱氨酸的天冬氨酸蛋白水解酶1(caspase-1,CASP1),使無活性CASP1前體(pro-CASP1)切割為有活性CASP1,從而促進無活性的IL-1β前體(pro-IL-1β)和IL-18前體(pro-IL-18)切割為成熟的IL-1β和IL-18,釋放到胞外參與機體炎癥及損傷等過程[1-3]。目前已發現的炎癥小體包括NLRP1、NLRP3、NLRP6、NLRP7、NLRP12、NLRC4以及NAIP[4]。本文就NLRC4炎癥小體的構成、活化機制以及其在感染和自身炎癥性疾病中的作用作一概述。
1 NLRC4炎癥小體的結構特征
NLRC4的LRR結構域由15個重復結構單元,共440個氨基酸組成。因每個結構單元之間與一個含有8~15個氨基酸的螺旋結構連接,故而被稱為富含亮氨酸的重復序列,該結構域用于識別病原相關分子模式(Pathogen associated molecular pattern,PAMP)等配體;NOD結構域屬于AAA+ATP酶的超家族,可介導NLRC4自身寡聚反應;CARD結構域通常為由NLRC4的前94個氨基酸組成,其折疊成六個反向平行的α-螺旋包裹在疏水核心周圍,可連接銜接蛋白ASC及效應分子CASP1,介導下游信號轉導。
Hu等[5]對在鼠巨噬細胞數量足夠、活性良好的情況下所分離出缺乏CARD結構域的NLRC4進行晶體分析,結果發現LRR、NOD結構域及占據NOD結構域的二磷酸腺苷(Adenosine diphosphate,ADP)通過廣泛的分子內相互作用以穩定該單體結構,進一步研究證實ADP近端關鍵殘基H443的突變可導致NLRC4寡聚化,從而促進炎癥小體的活化。研究者們發現人類的NLRC4功能突變大多集中在ADP結合位點附近,這表明核苷酸結合或ATP水解是寡聚化和炎癥小體活化所必需。同樣的,ATP磷酸結合環(P-Loop)突變導致NLRC4不能誘導CASP1激活[6]。
NLRC4寡聚化結構中存在單個Naip基因表達的結構,該基因表達產物可以將數個NLRC4分子組裝成盤狀炎癥小體,進而激活CASP1,產生一系列的免疫應答反應[7]。該盤狀炎癥小體較單一NLRC4結構活性更高,該活性主要通過在LRR與中央核苷酸結合結構域之間的聯合區域來體現。
2 NLRC4炎癥小體的活化機制
NLRC4的表達可通過腫瘤壞死因子(TNF-α)的刺激及p53活化而上調[8,9]。然而許多研究表明NLRC4表達的基礎水平已經足以支持NLRC4炎癥小體在上皮細胞和免疫細胞中的活化。
Mariathasan等[10]發現NLRC4缺失的鼠類巨噬細胞在接觸沙門氏菌后,CASP1不能被激活。隨后有多項研究表明來自沙門氏菌及軍團菌的鞭毛蛋白能夠誘導NLRC4介導的CASP1活化[11-13]。因此,研究者們認為鞭毛蛋白在NLRC4介導的CASP1活化過程中不可或缺。然而進一步的研究發現,雖然鞭毛蛋白缺失的沙門氏菌在感染早期不能激活CASP1,但在感染后期仍可監測到CASP1的活化和IL-1β的分泌。Miao等[14]發現此時CASP1的激活依賴于沙門氏菌Ⅲ型分泌系統(T3SS)的表達。因此,NLRC4的激活劑主要為鞭毛蛋白及T3SS蛋白(包括針狀蛋白分子和桿狀蛋白分子)。小鼠Naip基因家族分子中的Naip5、Naip6通過識別鞭毛蛋白介導NLRC4炎癥小體的激活,而Naip1及Naip2分別通過T3SS針狀蛋白分子和桿狀蛋白分子介導NLRC4炎癥小體的激活[15]。人類雖沒有與小鼠相同的Naip基因,但其表達的Naip同源物可通過識別T3SS針狀蛋白分子、桿狀蛋白分子和鞭毛蛋白激活NLRC4炎癥小體[16-18](圖1)。然而,細菌配體是如何結合Naip從而激活炎癥小體的機制尚不清楚。
為了研究NLRC4炎癥小體活化的具體機制,Qu等[19]采用人工遺傳學改造的小鼠進行實驗,研究發現沙門氏菌感染小鼠后可以誘導NLRC4第533位絲氨酸發生磷酸化修飾反應,即使抑制CASP1的活性也不能阻止這種磷酸化反應的發生,這說明NLRC4的磷酸化修飾反應位于CASP1切割的上游。此外,研究還發現蛋白激酶Cδ(Protein kinase Cδ,PKCδ)和p21活化激酶2(P21-activated kinase2,PAK2)都是參與NLRC4磷酸化修飾反應的磷酸激酶。Liu等[20]發現與帕金森癥和克羅恩病相關的富含亮氨酸重復序列激酶2(Leucine-rich repeat kinase 2 gene,LRRK2)表達減少可導致NLRC4炎癥小體活化程度下降。然而在幽門螺旋桿菌中鞭毛蛋白能夠誘導NLRC4磷酸化,但不能激活NLRC4炎癥小體[21]。因此,單純的磷酸化不足以誘導NLRC4的活化。NLRC4磷酸化相關激酶的特性及其在NLRC4活化過程中是否發揮著主導作用尚未清楚,但大量研究表明確保NLRC4的活化是抵御感染的關鍵部分。
3 NLRC4炎癥小體的效應機制
最初研究者們認為NLRC4通過激活CASP1,從而促進pro-IL-1β及pro-IL-18轉變為活性IL-1β及IL-18,引起炎癥反應。隨著研究的深入,我們發現NLRC4下游存在幾種不同的信號傳導途徑。CASP1還可以激活一種被稱為Gasdermin-D的胞質蛋白,該蛋白經切割后的N-末端結構域作用在宿主細胞膜上,導致細胞膜完整性喪失,并最終誘導細胞焦亡(Pyroptosis)[22]。與細胞凋亡不同,細胞焦亡的發生速度更快,IL-1β及IL-18等炎癥因子可通過Gasdermin-D形成的膜孔釋放到細胞外,促進機體抵御病原體(圖1)。除此之外,Moltke等[23]研究發現一種人造鞭毛蛋白衍生的NAIP5-NLRC4激活劑通過激活鈣依賴性磷脂酶A2(Phospholipase A2,PLA2)釋放花生四烯酸。通過CASP1活化動員的花生四烯酸促進前列腺素和白三烯的快速合成。CASP1動員花生四烯酸釋放的機制及該機制是否和NLRC4活化相關尚不清楚。目前,蛋白質組學研究已經確定了多達1 000個CASP1蛋白的潛在靶點,這表明除了上述信號傳導途徑外,可能還有其他的下游信號對NLRC4炎癥小體介導的CASP1激活產生應答。
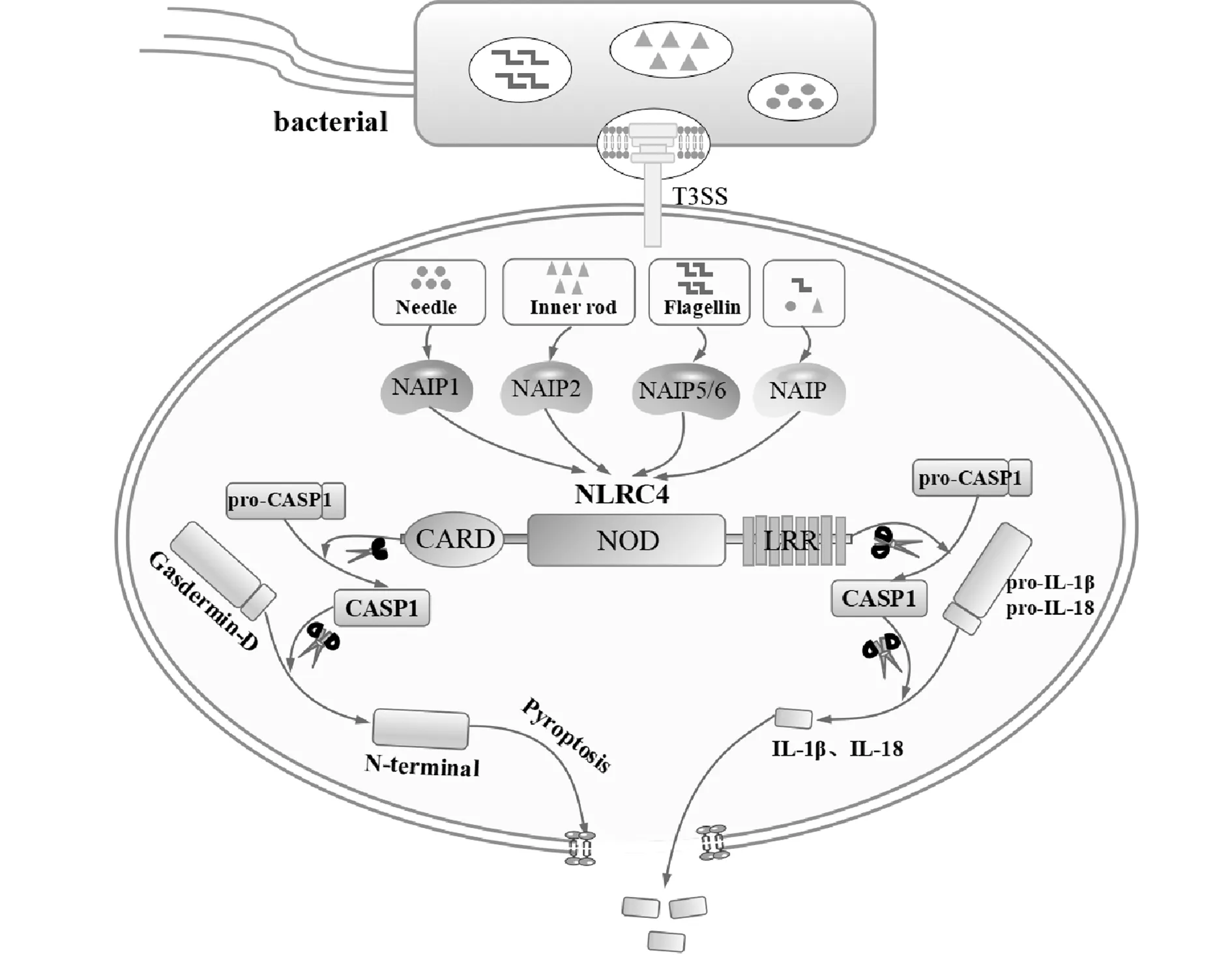
圖1 NLRC4激活與效應機制圖Fig.1 Triggering and effect mechanism of NLRC4Note:Four major groups of bacterial components trigger activation of the inflammasome.Flagellin and components of the type Ⅲ secretion system (T3SS) can be injected into the cytoplasm of the host cell via the T3SS.The T3SS needle and inner rod proteins are sensed by mouse NAIP1 and NAIP2,respectively,whereas flagellin is sensed by mouse NAIP5 or NAIP6.The needle and inner rod proteins and flagellin are all sensed by human NAIP.Ligand-bound NAIPs recruit NLRC4 to the same complex to drive activation of the NLRC4 inflammasome.NLRC4 acts as an adapter for caspase-1 activation.Caspase-1 cleaves the poreforming factor gasdermin D,whereby the N-terminal domain of gasdermin D forms pores in the host cell membrane.Caspase-1 also cleaves the proinflammatory cytokines pro-IL-1β and pro-IL-18,generating biologically active versions of these cytokines for release through the membrane pores generated by gasdermin D.The pores formed by gasdermin D also lead to lytic cell death via pyroptosis.
NLRC4的CARD結構域可直接與pro-CASP1相互作用從而催化CASP1激活,凋亡相關斑點樣蛋白(Apoptosis-associated speck-like protein,ASC)在這一激活過程中起正性作用。其他構成炎癥小體的蛋白(包括NLRP3,PYRIN和AIM2)亦可通過與ASC相互作用,進而募集并誘導pro-CASP1裂解成為有活性的P10/P20四聚體,促使pro-IL-1β及pro-IL-18切割成活性形式并釋放到細胞外參與免疫應答[24]。研究發現CASP8與CASP1類似,可促進pro-IL-1β轉變為活性IL-1β[25]。與CASP1不同的是,CASP8能在CASP1或Gasdermin-D被耗盡時引發CASP8依賴性細胞凋亡[26]。
4 NLRC4炎癥小體與鼠類宿主防御
Norlander等[27]發現感染檸檬酸桿菌后,Nlrc4-/-小鼠比野生型小鼠體重更輕,腸道炎癥的病理學特征(包括增生、白細胞浸潤和水腫)更明顯。另外,有實驗通過給予僅在腸道上皮細胞中特定表達NLRC4蛋白的實驗小鼠注射純化后的細菌鞭毛毒素(Fla Tox),結果發現相對于不表達NLRC4蛋白的老鼠,表達NLRC4蛋白的小鼠炎癥反應更重,細菌數要更少[28]。Sellin等[29]證實小鼠腸上皮細胞Naip1-6組織特異性缺失可使感染早期沙門氏菌細菌負荷增加,這表明將沙門氏菌感染的腸上皮細胞排出到腸腔中取決于腸上皮細胞NAIP蛋白的表達。以上研究表明NAIP-NLRC4炎癥小體在控制腸道感染的細菌病原體中發揮了重要的作用。
盡管對于NLRC4的研究主要集中在腸道感染及腸道病原體上,近年研究發現NLRC4也與非腸道感染相關。產單核細胞李斯特菌的鞭毛蛋白可激活NLRC4,從而誘導細胞死亡[30]。Balakrishnan等[31]發現機體在感染銅綠假單胞菌后,可通過T3SS組分激活NLRC4炎癥小體,引發肺部炎癥反應。而且NLRC4可以抵御非鞭毛桿菌如肺炎克雷伯菌[32]。這些非腸道病原體通過類似于沙門氏菌和志賀氏菌等腸道病原體激活NLRC4的典型機制(通過鞭毛蛋白或細菌T3SS的組分)激活NLRC4。此外,一些病原體(如立克次體)可通過環氧合酶2(Cyclooxyg-enase-2,COX2)介導的前列腺素合成的機制誘導巨噬細胞NLRC4活化[33]。
5 NLRC4炎癥小體與人類自身炎癥性疾病
NLRC4在鼠類宿主防御機制中發揮著重要的作用,前文提及人Naip可通過識別T3SS針狀蛋白分子激活NLRC4炎癥小體,我們推測NLRC4在人類細胞內的細菌感染及清除中也扮演著不可或缺的角色。然而,關于NLRC4在現代人類機體中清除病原體的相關研究仍待進一步完善,且目前尚無人類NLRC4功能喪失的相關研究。
研究者們發現NLRC4在驅動人類自身炎癥性疾病(Autoinflammatory disease,AID)中也發揮著重要作用。自身炎癥是指不歸因于感染、惡性腫瘤或抗原特異性自身免疫的全身或器官特異性炎癥[34],其最初范疇是圍繞自發性或增強的炎癥小體活化相關的可以阻斷IL-1β的單基因疾病,自1999年McDermott等[35]提出后,這個概念現已擴展為包括數量驚人的疾病,其中許多是因為不適當的炎癥小體激活所致。
經過10余年對PYRIN和NLRP3炎癥小體以及相關自身炎癥性疾病如家族性地中海熱(Familial mediterranean fever,FMF)和冷炎素相關周期熱綜合征(Cryopyrin-associated periodic syndromes,CAPS)的深入研究,NLRC4相關自身炎癥性疾病于2014年被首次提及。研究表明NLRC4基因功能獲得性突變所導致自發/增強炎癥小體的異常活化是引起小兒小腸結腸炎和復發性巨噬細胞活化綜合征(Macrophage activation syndrome,MAS)的原因[36,37]。
MAS是一種以急性發熱、全血細胞減少、肝膽功能障礙、凝血功能障礙及血清鐵蛋白持續升高為特征的綜合征[38],臨床上類似于致命的嗜血細胞性淋巴組織細胞增多癥,通常被歸類于免疫缺陷。其是兒童慢性風濕性疾病的嚴重并發癥,通常使風濕性疾病特別是全身型幼年特發性關節炎(systemic juvenile idiopathic arthritis,sJIA)和成人斯蒂爾病(Adult onset still′s disease,AOSD)復雜化。盡管IL-1β和IL-18在骨髓細胞中表達并在活化的炎癥小體參與下成熟并釋放到胞外,但在發現NLRC4-MAS之前,感染性疾病患者的血清中并未檢測到大量的IL-1β和IL-18。相反在sJIA和AOSD,尤其是MAS患者中可檢測到極高水平的IL-18[39,40]。因此,盡管NLRC4與感染性疾病及MAS均相關,但IL-18的高水平似乎只與后者相關。且患有難治性重癥MAS的患者在使用IL-18阻斷劑及γ干擾素阻斷劑(一種與IL-18誘導產生的細胞因子)后療效較好[41]。這表明IL-18可能是炎癥性疾病的潛在靶標。
不同于其他炎癥腸病,NLRC4相關小腸結腸炎病例均發生在嬰兒早期,其炎癥可波及從胃到結腸的整個腸道,病變發生在十二指腸的患者癥狀較輕。NLRC4相關小腸結腸炎患者的腸道活組織檢查和尸檢結果均表現為混合的炎癥浸潤,伴有組織水腫、上皮糜爛和組織自溶[42,43]。目前研究發現該病的發生與NLRC4基因突變相關,常見的突變位點為:V341A、T337S、T337N和S171F[33,42,44,45]。第337和341位氨基酸均位于炎癥小體NOD結構域中,NLRC4晶體分析表明第341位氨基酸在ADP轉化為ATP的過程中發揮著重要的作用,其突變可影響炎癥小體的活化過程[42]。337位氨基酸可通過與170位和173位相互作用以穩定炎癥小體的ADP結合位點結構[33]。據推測,用苯丙氨酸代替171位絲氨酸可改變上述相互作用,從而影響炎癥小體活化,導致NLRC4相關小腸結腸炎發生。令人震驚的是盡管結腸炎嬰兒伴發MAS、血清IL-18水平維持在較高水平,但能從嬰兒期存活下來的患者的胃腸疾病可以痊愈[36,37]。因此,早期腸道定植可能會促進NLRC4相關小腸結腸炎患者細胞因子產生,該疾病的自然消退可能與腸道成熟、腸道黏膜免疫及腸道菌群相關。NLRC4在腸道防御中的作用提示NLRC4和IL-18與腸道微生態及免疫穩態之間存在復雜的相互作用。
除了MAS和小腸結腸炎,研究發現在患有FACS4(一種自身炎癥性疾病)的兒童體內存在NLRC4突變體H443P[46]。Volker-Touw等[47]發現另一多種自身炎癥表型的家系(包括蕁麻疹和結節性皮疹、結膜炎、關節炎等)也與NLRC4的錯義突變相關。血清IL-18在該家系成員中顯著升高。Kawasaki等[48]記錄了一位具有嚴重冷炎素相關周期熱綜合征臨床癥狀的患者,該患者具有新型NLRC4突變的體細胞嵌合體。因此,NLRC4與自身炎癥性疾病具有明確的聯系。NLRC4激活對類花生酸生成的特異性尚未在這些患者中進行評估,但抑制環氧合酶在除輕度患者以外的所有治療中都療效不佳。
大量研究表明NLRC4突變的氨基酸位置與自身炎癥性疾病表型之間存在一定的相關性。Unal等[49]發現在1例患有sJIA和復發MAS的患兒中,其發生了W374X突變。H443P突變可增加NLRC4與SUG1和泛素化蛋白質的相互作用,并最終在人肺上皮細胞系中驅動CASP8依賴性細胞凋亡[50]。發生在核苷酸結合域(Nucleotide binding domain,NBD)內的第171位及第177位突變分別導致子宮復發性巨噬細胞活化綜合征,胎盤血栓形成及嚴重冷炎素相關周期熱綜合征,迄今為止公布的所有突變都發生在ADP/ATP結合位點附近[5]。目前,對于NLRC4參與免疫調控的機制研究主要集中在利用小鼠基因敲除模型證明NLRC4在各種細菌中激活CASP1的作用。NLRC4及相關信號通路在sJIA和AOSD等常見疾病及宿主防御方面的機制仍待進一步研究及驗證。
6 小結
盡管NLRC4炎癥小體相關的研究開始較晚,但目前研究已為炎癥小體和自身炎癥疾病的關系提供了較為確切的依據。關于鼠NLRC4活性的微生物學和細胞免疫學的研究已經證實NLRC4在固有免疫宿主防御機制中的重要作用,并確定了其作用位點的調節途徑、調節機制(如磷酸化)、相互作用的對象(例如其他NLRs、CASP8)和效應機制。為指導人類炎癥性疾病的預防和治療,NLRC4與人類炎癥性疾病的關聯,尤其是其與IL-18、MAS和嬰兒小腸結腸炎的獨特關聯需要更進一步的研究。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
