基于動態(tài)多反應監(jiān)測模式下的高效液相色譜-串聯(lián)質譜法同時測定枸杞中44種農(nóng)藥殘留
2020-02-18 08:21:24
食品工業(yè)科技 2020年1期
(蘭州市食品藥品檢驗所,甘肅蘭州 730050)
枸杞(LyciumbarbarumL.)是一種重要的藥食同源農(nóng)產(chǎn)品,深受廣大消費者的喜愛[1]。枸杞因多適宜在鹽堿地和退耕還林地種植,使其成為西北地區(qū)主要經(jīng)濟作物[2],主要分布在寧夏、甘肅、新疆、青海等地。由于其含糖量高,病蟲害情況嚴重,目前主要是以化學藥物防治[3],但生產(chǎn)者為了確保產(chǎn)量,常常對其不規(guī)范使用化學藥物,造成了農(nóng)藥殘留超標,這不僅對生態(tài)環(huán)境造成污染,更可能對消費者的身體健康產(chǎn)生直接或者潛在的危害。因此,建立枸杞中農(nóng)藥殘留的檢測方法對于保障枸杞市場的質量和人們的健康具有重要的意義。
目前,枸杞中農(nóng)藥殘留儀器檢測技術有很多,主要是氣相色譜法[4]、氣相色譜-串聯(lián)質譜法[5-6]、液相色譜法[7]、液相色譜-串聯(lián)質譜法[3,8-10]等。其中高效液相色譜-串聯(lián)質譜法是一種具有靈敏度高、分析速度快、高通量等特性的定性定量分析方法,在多種農(nóng)藥殘留分析中被廣泛應用。目前,利用高效液相色譜-串聯(lián)質譜法測定農(nóng)藥殘留的監(jiān)測模式多數(shù)是采用多反應監(jiān)測模式,對動態(tài)多反應監(jiān)測模式的檢測還鮮有報道。羅輝泰[11]在測定飼料中藥物殘留采用了動態(tài)多反應監(jiān)測方式,在保證準確定量的情況下仍具有較高的檢測靈敏度,但是關于枸杞中農(nóng)藥殘留采用動態(tài)多反應監(jiān)測方式進行測定的至今尚未見報道。動態(tài)多反應監(jiān)測模式的優(yōu)勢是根據(jù)色譜分析時間對每個組分的保留時間窗口進行動態(tài)分配,僅當組分流出時才被采集,這種科學的分段方式有效減少了同時掃描不同的離子對的數(shù)目,并一定程度上提高了駐留時間,大大提高了整個實驗的分析效率。本實驗的研究對象枸杞干果含水量低,且含有多糖、有機酸、類胡蘿卜素、脂肪等物質[12],在測定時需要充分提取和凈化,因此本實驗考慮從復水量、提取溶劑、凈化填料和配比等條件優(yōu)化來提高提取凈化效果,通過高效液相色譜-串聯(lián)質譜法的動態(tài)多反應監(jiān)測模式測定建立簡單快速、準確可靠的同時測定44種農(nóng)藥多殘留分析方法,為枸杞中多種農(nóng)藥殘留快速檢測提供科學的技術支持。
1 材料與方法
1.1 材料與儀器
枸杞 蘭州市售;甲胺磷、乙酰甲胺磷、氧樂果、霜霉威、多菌靈、噻菌靈、滅多威、噻蟲嗪、吡蟲啉、樂果、啶蟲脒、硫環(huán)磷、抑霉唑、甲基硫菌靈、甲萘威、嘧霉胺、異丙威、烯酰嗎啉、乙霉威、丙森鋅、嘧菌酯、啶酰菌胺、氟硅唑、戊菌唑、聯(lián)苯肼酯、除蟲脲、蟲酰肼、氯唑磷、甲氨基阿維菌素苯甲酸鹽、苯霜靈、氰霜唑、苯酰菌胺、四螨嗪、辛硫磷、氟苯脲、噻嗪酮、炔螨特、噠螨靈、涕滅威 均為德國Dr. Ehrenstorfer. Gmbh公司;殺線磷、久效磷、甲基硫環(huán)磷 均為北京曼哈格;氯吡脲、噻螨酮 均為北京鄭翔科技有限公司;以上對照品純度均≥98.0%。乙腈、甲醇、甲酸、乙酸 均為色譜純,德國Merck公司;乙酸銨 HPLC級,美國Sigma公司;丙酮 色譜純,科密歐化學試劑有限公司;QuEChERS凈化管填料:乙二胺-N-丙基硅烷化硅膠(PSA)40~60 μm、石墨化炭黑(GCB)40~120 μm、十八烷基硅烷鍵合硅膠(C18)40~60 μm 均為 美國Agilent 公司;無水硫酸鎂、無水醋酸鈉、氯化納、檸檬酸二鈉、檸檬酸鈉 均為分析純,國藥集團化學試劑有限公司。
1260HPLC-6460三重四極桿液質聯(lián)用儀 美國Agilent公司;5810R高速低溫離心機 德國Eppendorf公司;A11基本型分析研磨機,KS501圓周振蕩搖床 德國IKA公司;MS205DU電子天平、BSA224S-CW電子天平 德國Sartorius公司;EVA32多功能樣品濃縮儀 北京普立泰科公司;VORTEX-5渦旋混勻器 海門其林貝爾公司;Milli-Q超純水機 美國Millipore公司。
1.2 實驗方法
1.2.1 標準溶液配制 準確稱取各供試標準品,據(jù)標準物質溶解性用乙腈、甲醇分別制成1.00 mg/mL的標準儲備液,4 ℃冷藏避光保存,備用;臨用前配制成0.002~0.200 mg/L的44種混合標準工作溶液。
1.2.2 樣品制備 取枸杞干果樣品,-20 ℃冷凍48 h后立即用分析研磨機粉碎,精密稱取樣品5 g(精確至0.001 g),置50 mL離心管中,加入10 mL超純水振蕩混勻(復水處理),再加入10 mL 0.1%甲酸-乙腈,渦旋1 min,在搖床上振蕩20 min,放入冰箱中-18 ℃冷凍20 min,加入4 g硫酸鎂、1 g氯化鈉、1 g檸檬酸鈉、0.5 g檸檬酸氫二鈉,立即渦旋混合均勻1 min,以4 ℃、3900 r/min離心10 min,取上清液,待凈化。精密移取5.00 mL上清液加到內(nèi)含PSA 150 mg、C18150 mg、MgSO4900 mg的15 mL塑料離心管中,渦旋混勻1 min,4 ℃、3900 r/min離心10 min,精密量取2.00 mL上清液于7 mL聚乙烯離心管中,40 ℃水浴中氮吹至近干,加入2.00 mL乙腈-水(3∶2)溶液復溶,過0.2 μm濾膜后上機測定。
1.2.3 基質匹配標準溶液的配制 依照1.2.2方法制備樣品提取液,以此基質溶液配制成基質匹配標準使用液。
1.2.4 儀器條件 液相色譜條件:色譜柱Water Acquity HSS T3(2.1 mm×100 mm,1.8 μm),進樣量5 μL,柱溫30 ℃,流速0.3 mL/min,后運行時間5 min,梯度洗脫程序見表1。

表1 流動相梯度條件Table 1 Gradient elution process for liquid phase
質譜條件:電噴霧離子源ESI,動態(tài)多反應監(jiān)測(d-MRM),正離子模式,霧化氣壓力40 psi,干燥器溫度350 ℃,干燥氣流速10 L/min,毛細管電壓4000 V,腔電流:0.28 μA。質譜條件見表2。

表2 44種農(nóng)藥的質譜條件Table 2 Mass spectrometric parameters of 44 kinds of pesticides
1.3 數(shù)據(jù)處理
采用MassHunter Quantitative Analysis軟件處理數(shù)據(jù),以MassHunter Quanlitative Analysis和Microsoft Excel 2007軟件分析并繪圖。
2 結果與分析
2.1 色譜條件的優(yōu)化
2.1.1 流動相梯度洗脫條件的確定 以0.1%甲酸水(a)和乙腈(b)為流動相,比較了方案A(0~2 min,90% a;2.1~5 min,90% a→50% a,5~15 min,50% a;15~15.1 min,90% a;15.1~20 min,90% a)和方案B(表1)兩種梯度條件下的44種農(nóng)藥d-MRM色譜圖(見圖1)。從圖1可以看出,方案A出峰時間相對集中,峰的重疊會影響各個目標化合物的峰形及其分離效果,導致響應降低。方案B各個化合物依次出峰,分離效果好,峰形改善,響應提高。因此,選擇方案B為本實驗的最佳梯度洗脫條件。

圖1 兩種梯度洗脫方案對44種農(nóng)藥出峰的影響Fig.1 Effects of two kinds of gradient elution schemes on the peak of 44 kinds of pesticides
2.1.2 流動相種類的確定 比較了A:乙腈-水,B:乙腈-0.1%甲酸水溶液,C:乙腈-0.1%乙酸水溶液,D:乙腈-0.1%甲酸2 mmol/L乙酸銨的水溶液4種流動相體系下的色譜圖(圖2)。從圖2可以看出,同等測定條件下,流動相體系C下各個目標化合物的響應偏低。當流動相為A、B、D時,從峰的分離度看,在3~5.5 min時間段內(nèi),A中色譜峰重疊較多,D中色譜峰響應偏低;流動相A在9~10 min內(nèi)抑霉唑和甲基硫菌靈未完全分離;11~12min內(nèi)A中的三種化合物全部分離,B和D中的嘧霉胺和氯吡脲重疊;從色譜峰的整體響應來看,響應強度順序為B>A>D。因此,綜合考慮峰的分離效果和響應情況,選擇最佳的流動相種類為乙腈-0.1%甲酸水溶液。

圖2 流動相種類對44種農(nóng)藥出峰的影響Fig.2 Effects of mobile phase species on the peak of 44 kinds of pesticides
2.2 質譜條件的優(yōu)化
將44種農(nóng)藥的單個標準溶液(0.1 μg/mL)采用自動優(yōu)化和手動優(yōu)化兩種方式對其質譜條件分別進行確定,然后建立全段MRM方法采集44種農(nóng)藥化合物名稱和保留時間等信息,利用獲得的已知信息轉化為動態(tài)多反應監(jiān)測模式(d-MRM),d-MRM的建立可避免不分段采集方式中多個MRM重疊使靈敏度降低的難題,以及分段采集方式中相鄰峰在分段處保留時間漂移需在兩個段中均設置它們的MRM所帶來的降低分析方法通量的缺陷,d-MRM適合多種農(nóng)藥的同時檢測,不僅可顯著改善多種化合物負載循環(huán)時間,還可提高靈敏度和選擇性。因此,本文以d-MRM方式建立了44種農(nóng)藥的質譜條件。
2.3 復水量的優(yōu)化
枸杞干果含水量低,直接用乙腈提取難以充分滲透到樣品組織內(nèi)部,經(jīng)復水處理后再提取會改變提取效率,因此,比較了10、15、20 mL的復水量對44種農(nóng)藥的提取效果(見圖3)。從圖3結果可以看出,除噻蟲嗪、吡蟲啉、甲基硫菌靈和氟苯脲的回收率整體偏低外,當復水量為10 mL時,甲胺磷、乙酰甲胺磷和氧樂果的回收率低于復水量15、20 mL外,其它化合物的回收率范圍為64%~119%,從整體回收率水平來看,復水量10、15、20 mL的平均回收率為87.7%、61.9%、61.2%,可能是由于復水量太大,枸杞中的一部分糖類物質溶入水中進而影響凈化效果,使得回收率降低。因此,本實驗得到最佳的復水量為10 mL。采用液質聯(lián)用測定枸杞中農(nóng)藥殘留,梁月香[13]選擇5 g樣品的最佳復水量為5 mL;GB 23200.11-2016[14]中規(guī)定2 g的加水量為5 mL;本實驗中5 g樣品的最佳復水量是10 mL,可見,最佳復水量的選擇可能與供試樣品的干燥程度、含糖量等因素有關。
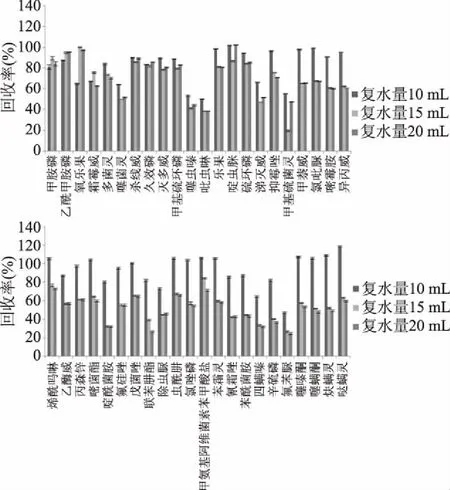
圖3 不同復水量對44種農(nóng)藥化合物的提取效果的影響Fig.3 Effect of different water addition on the extraction efficiency of 44 kinds of pesticide compounds
2.4 提取劑的優(yōu)化
選取乙腈、0.1%甲酸-乙腈、0.1%乙酸-乙腈、乙酸乙酯為提取劑,通過比較目標化合物的回收率確定最佳提取溶劑(圖4)。結果顯示,乙酸乙酯對各種化合物的提取回收率在50%左右,提取效果較低。比較乙腈、0.1%甲酸-乙腈、0.1%乙酸-乙腈提取劑對目標化合物的回收率發(fā)現(xiàn):3種條件下甲基硫菌靈、氧樂果和氟苯脲的回收率均偏低。與乙腈和0.1%乙酸-乙腈相比,0.1%甲酸-乙腈條件下上述化合物的提取回收率較好,且其它化合物的回收率均在60%~120%之間。乙腈、0.1%甲酸-乙腈、0.1%乙酸-乙腈提取條件下的平均回收率分別為87.79%、88.97%、86.49%,因此,本實驗選取的最佳提取溶劑為0.1%甲酸-乙腈。

圖4 不同提取劑對44種農(nóng)藥化合物的提取效果的影響Fig.4 Effect of different solvent on the extraction efficiency of 44 kinds of pesticide compounds
2.5 凈化條件的優(yōu)化
以3種不同填料和配比的QuEChERS凈化管為凈化方案(A:PSA 150 mg、C18150 mg、MgSO4900 mg,B:PSA 150 mg、GCB 15 mg、MgSO4885 mg,C:PSA 150 mg、GCB 45 mg、MgSO4855 mg)對44種農(nóng)藥提取液進行凈化處理,通過比較化合物的回收率確定最佳凈化方案(圖5)。從圖5可以看出,方案C中各個化合物的回收率不均衡,除了甲胺磷、乙酰甲胺磷、氧樂果、霜霉威的回收率達到80%以上,其它化合物的回收率均較低,無法達到試驗要求。方案A和B各個化合物的回收率均在60%~118%之間,平均回收率分別為87.4%和84.5%,均能滿足試驗要求。PSA主要用于除樣品中的極性有機酸和糖類,C18主要用于除去樣品中的油脂和類胡蘿卜素,GCB主要去除葉綠素等色素,且在萃取極性化合物的回收率較C18更高更穩(wěn)定[15],方案B中甲胺磷、乙酰甲胺磷等極性化合物的回收率分別為69.8%、80.7%,較方案A的68.7%和79.3%略高;而其余化合物的回收率均低于方案A。綜合考慮,本實驗以方案A的填料和配比為凈化條件。

圖5 不同凈化條件對44種農(nóng)藥化合物凈化效果的影響Fig.5 Effect of different purification conditions on the extraction efficiency of 44 kinds of pesticide compounds
2.6 基質效應的影響
液相色譜-串聯(lián)質譜法測定農(nóng)藥殘留常常會受到樣品基質的干擾,本文將枸杞基質溶液和乙腈水溶液分別制備成0.02、0.08、0.16 μg/mL的基質標樣和溶劑標樣,以基質標樣的響應與溶劑標樣響應的比值得到基質效應(ME)。當ME>1時,說明基質的存在增強了分析物的響應;當ME<1,則基質的存在抑制了分析物的響應;當ME=1,表示不存在基質效應[16]。結果表明,多數(shù)農(nóng)藥呈基質減弱效應,濃度越低,基質減弱效應越明顯,尤其是極性強的部分農(nóng)藥,當濃度為0.02 μg/mL時,甲胺磷、噻蟲嗪和吡蟲啉的基質效應為0.75~0.87,其它化合物的基質效應不太明顯,在0.90~1.01之間。隨著農(nóng)藥濃度的升高,部分農(nóng)藥反而呈現(xiàn)輕微的基質增強效應。因此,在今后的枸杞農(nóng)藥殘留檢測中,對于那些基質效應強的農(nóng)藥,建議最好選擇基質匹配標曲法進行定量分析[17],對于基質效應不明顯的農(nóng)藥,可以用溶劑標曲法定量分析。
2.7 方法學驗證
2.7.1 線性方程及檢出限、定量限 44種農(nóng)藥在0.002~0.200 mg/L范圍內(nèi)具有良好的線性關系(R2>0.9943),以儀器的3倍和10倍信噪比(S/N)表示方法的檢出限和定量限,分別為0.01~3.30和0.03~11.0 μg/kg,均能滿足農(nóng)藥殘留分析要求。

表3 44種農(nóng)藥化合物的線性關系、檢出限和定量限Table 3 Linear relationship,LODs and LOQs for 44 kinds of pesticide compounds
2.7.2 回收率及重復性試驗 在枸杞空白樣品中分別添加低、中、高(0.04、0.08、0.16 mg/kg)3個濃度水平的加標回收率試驗,每個濃度3次平行實驗,低、中濃度每個平行重復測定3次,高濃度平行測定6次,各個化合物的加標回收率和相對標準偏差結果見表4。結果顯示,44種農(nóng)藥的回收率范圍為60.2%~115.3%,相對標準偏差為0.63%~8.27%。部分檢驗項目的回收率偏低,可能是枸杞基質效應的影響、農(nóng)藥自身極性和穩(wěn)定性的特點、混標溶液間存在的干擾等多方面因素造成。

表4 44種農(nóng)藥的回收率和相對標準偏差Table 4 Average recoveries and RSDs of 44 kinds of pestcides
2.8 實際樣品的測定
在市場隨機購買樣品20批,共檢出農(nóng)藥14種(見表5)。其中多菌靈、啶蟲脒、吡蟲啉的檢出率較高,均達到95%以上;烯酰嗎啉和四螨嗪的檢出率次之,分別為75%和60%。
從測定結果還可以看出,14種農(nóng)藥的殘留水平值均較低,啶蟲脒遠遠低于GB 2763-2016[18]規(guī)定的最大殘留限量值1 mg/kg。本實驗中檢出率高的三種農(nóng)藥,與王瑩[19]和蔣玉寶[20]報道的結果相一致。
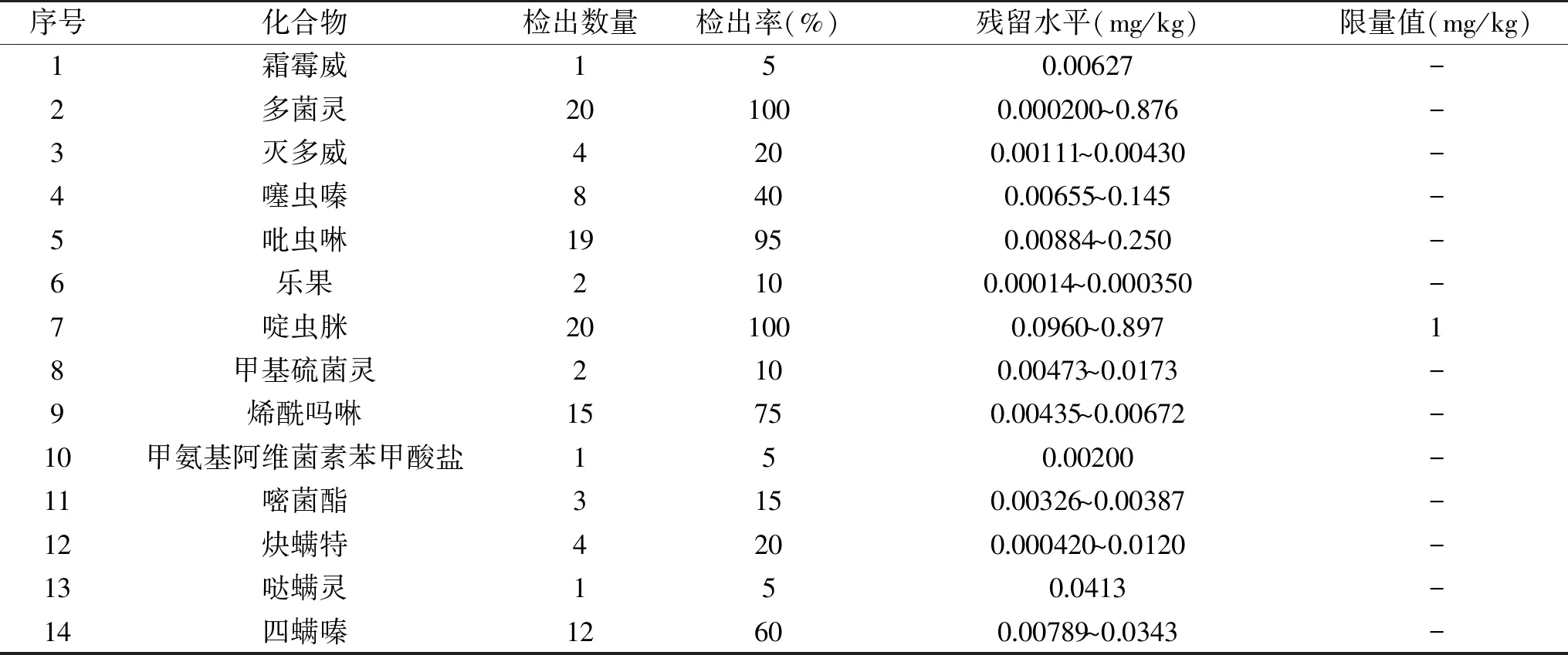
表5 實際樣品的測定Table 5 Determination of actual samples
3 結論
本實驗采用0.1%甲酸-乙腈提取,PSA 150 mg、C18150 mg、MgSO4900 mg分散固相萃取凈化,經(jīng)T3色譜柱分離,0.1%甲酸水和乙腈流動相體系梯度洗脫,采用電噴霧的動態(tài)多反應監(jiān)測模式檢測,基質匹配標準曲線的外標法定量,建立了同時測定枸杞中44種農(nóng)藥殘留的高效液相色譜-串聯(lián)質譜的分析檢測方法。結果表明,各農(nóng)藥在濃度范圍內(nèi)的線性關系良好,決定系數(shù)均大于等于0.9943,三個添加水平的回收率范圍為60.2%~115.3%,相對標準偏差為0.63%~8.27%,檢出限范圍為0.01~3.30 μg/kg。本方法操作簡單,準確度和精密度高,適合枸杞中多種農(nóng)藥殘留的定性和定量檢測,可為農(nóng)藥殘留的監(jiān)控和風險評估提供了一種快速高效的分析手段。
