乏燃料后處理中的輻射化學問題Ⅱ.水溶液和稀釋劑的輻射分解
2020-03-05 09:36:14翁漢欽葉國安林銘章
核化學與放射化學 2020年1期
湯 嘉,翁漢欽,何 輝,葉國安,林銘章,3,*
1.中國科學技術大學 物理學院 工程與應用物理系,安徽 合肥 230026;2.中國原子能科學研究院 放射化學研究所,北京 102413;3.中國科學院 核能安全技術研究所,安徽 合肥 230031
自1952年12月美國實現增值堆1號(EBR-1)首次利用核能發電以來,世界核電已有60多年的發展歷史。2014年,核電站向全球供應2 441 TWh的電能,這占到當年全球發電量的10%,而中國為了迎合能源結構改革,將大力發展核電。發展迅猛的核電行業帶來了經濟利益的同時,也隨之產生大量的乏燃料。乏燃料能否得到安全有效的處理、處置關系著核電的發展。為了實現核能的可持續發展戰略,我國采取對乏燃料進行閉式循環的戰略,即對乏燃料進行后處理,回收鈾、钚,并通過再循環加以利用,以提高核燃料的利用率,減少放射性產物的毒素和體積[1]。
關于乏燃料后處理的元素分離,已發展出多種商用和實驗室階段的后處理流程,如PUREX流程[2]、UREX流程[3]、COEX流程[4]、CSEX流程[5]、SREX流程[6]、FPEX流程[7]、TRUEX流程[8]、DIAMEX流程[9]、ARTIST流程[10]、TALSPEAK流程[11]、GANEX流程[12]。由于乏燃料具有放射性的特殊性,這些濕法流程中水相和有機相不可避免會受到238Pu、241Am等α輻射源產生的α輻射以及裂片核素如90Sr、137Cs產生的β和γ輻射。該過程中產生的輻射使得水相和有機相的物質發生化學反應。簡而言之,在乏燃料后處理中需要關注輻射化學的主要原因在于:(1) 輻射分解降低了配體濃度從而導致萃取效率降低;(2) 輻解產物往往是新的絡合劑從而導致分離因子降低;(3) 輻解產物經常是親水性的絡合劑,因而對反萃不利;(4) 輻射分解產生的中間活性粒種可能改變金屬的價態。
本綜述系列Ⅰ[13]已經對磷酸三丁酯(TBP)及多種新型的萃取劑的輻射穩定性進行了介紹和討論。本文作為系列Ⅱ,將主要從硝酸輻解、錒系水溶液化學、稀釋劑的輻射穩定性以及乏燃料的氧化溶解四個方面介紹,并簡單解釋輻解的機理。
1 硝酸的輻解
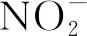
研究硝酸輻解的途徑主要包括穩態輻解和脈沖輻解兩種方法。隨著技術的進步以及研究的深入,時間尺度更小的脈沖輻解裝置被研制出來[22],為硝酸輻解機理進一步的研究提供了基礎條件。
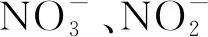
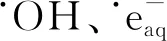

(1)
(2)
(3)
(4)
(5)
(6)
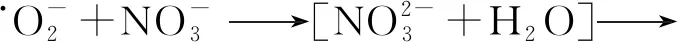
(7)
Garaix等[23]通過13.5 MeV的α輻射對含有NaNO3和HNO3的水溶液進行研究,進一步研究H2O2輻射化學產額,并給出了H2O2輻射化學產額(G(H2O2))的經驗公式(式(8)—(10))。
(8)
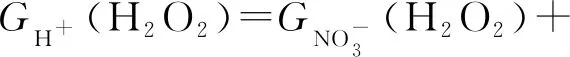
(9)
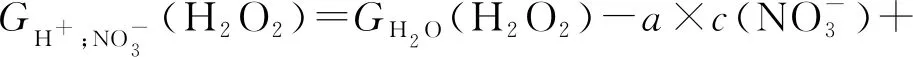
(10)

(11)
加入該機理反應進行模擬后,并在反應速率常數為1.0×1013dm3/(mol·s)時,對于硝酸影響水輻解的G(H2)的模擬結果與文獻[25-34]實驗數據一致。
Liu等[20]以238Pu作為輻射內源探究了水溶液硝酸的輻解過程,并給出了HNO2產生和消耗的關鍵方程式(12)—(17)。
(12)
(13)
(14)
(15)
(16)
(17)
式(12)—(17)表明·NO2、·NO和H2O2是HNO2產生的關鍵中間產物。
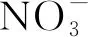
(18)
GH+(HNO2)=B×(1-exp(-g×c(H+)))
(19)
(20)
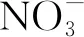
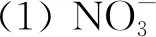
(21)
(22)
(23)
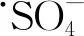
(24)
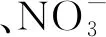
目前對·NO3生成機理的研究已經比較全面[18,46],有學者[49-52]對·NO3的消耗也開展了一定的工作,綜合認為·NO3有如下輻解機理(式(25)—(32)),Garaix等[53]通過ELYSE皮秒脈沖輻解裝置測定了·NO3輻解過程的化學反應速率常數(k)。
(25)
k=4.0×106L/(mol·s)
(26)
k=4.4×109L/(mol·s)
(27)
k=2.0×108L/(mol·s)
(28)
k=1.1×109L/(mol·s)
(29)
k=3.0×102L/(mol·s)
(30)
k=1.0×1010L/(mol·s)
(31)
k=3.0×109L/(mol·s)
(32)
k=7.1×106L/(mol·s)
Garaix等[53]還通過ELYSE皮秒脈沖輻解裝置探究酸度和肼對·NO3輻解的影響,結果表明:在中性無肼條件下,輻解產生的·NO3主要與四種物質發生反應,在單次脈沖為38 Gy的輻射條件下,·NO3主要由·NO2(式(28))、·NO3(式(26))、·OH(式(30))和H2O(式(29))消耗。在純7 mol/L HNO3溶液、單次脈沖為38 Gy的輻射體系下,·NO3主要和兩種物質反應:·NO2(式(28))和HNO2(式(27)),其中80%的·NO3由·NO2消耗,15%的·NO3由HNO2消耗。
由于實際后處理過程中,會引用氧化性物質如N2H4等抑制HNO2的產生,因此Garaix等[53]還研究了7 mol/L HNO3溶液加入N2H4后的·NO3的產額和消耗,發現N2H4的存在可以極大加速·NO3的消耗,機理在于式(33)[54-55]:
(33)
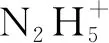
(34)
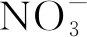
(35)
(36)
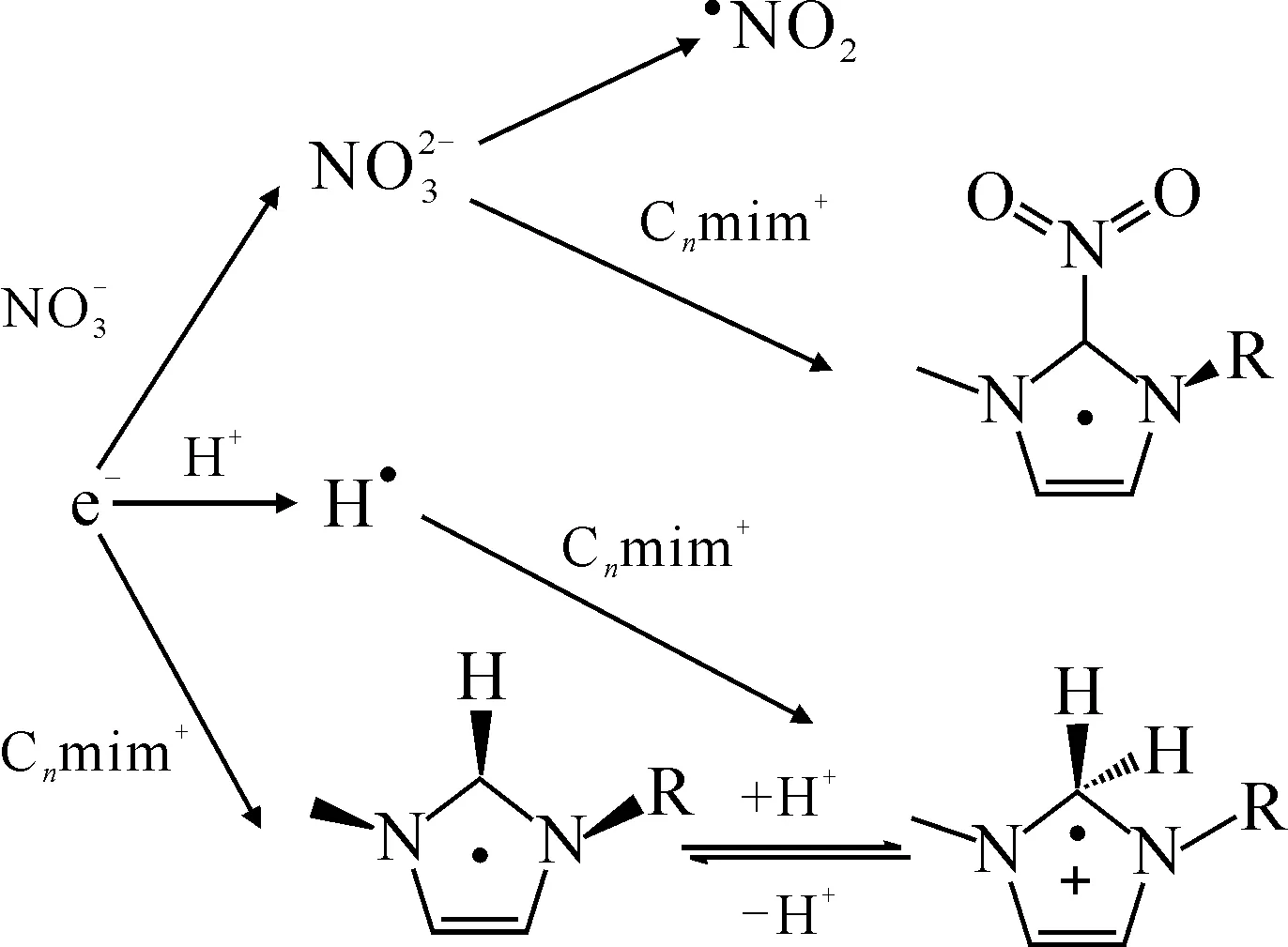
圖1 離子液體(陽離子為Cnmim+)輻解體系中硝酸清除e-的機理[56]Fig.1 Mechanism of removal of e- by nitric acid in irradiated ionic liquids(cations: Cnmim+) [56]
2 錒系水溶液的輻射化學
在后處理流程中,錒系元素的價態一定程度上決定了萃取分離的效率。而在電離輻照條件下,硝酸水溶液會產生還原性和氧化性的自由基等產物,從而影響金屬離子價態的變化。因此研究錒系水溶液輻射化學有實際指導意義。
Mincher等[58-59]比較了γ輻解條件下硝酸體系中Am(Ⅵ)和Np(Ⅴ)的氧化還原反應。Am(Ⅵ)通過NaBiO3氧化Am(Ⅲ)得到。對Am(Ⅵ)的硝酸溶液進行γ輻解,在0.3 kGy的累積輻射過程中,間斷性使用紫外/可見光譜采集數據,分析發現Am(Ⅵ)和Am(Ⅲ)的濃度隨著輻照劑量的增加逐漸降低,Am(Ⅴ)的濃度則隨輻照劑量的累積上升逐漸增加,這表明Am(Ⅵ)最終轉化為Am(Ⅴ)。Am(Ⅵ)轉化為Am(Ⅴ)的原因可能是由于持續的輻解,使得部分HNO3和H2O生成HNO2和一些還原性的自由基,這一推斷和Grimes等[60]的實驗論證不謀而合。Grimes等[60]探究了Am(Ⅵ)在HNO3水溶液中自動還原的動力學,給出了Am(Ⅵ)還原的機理(如式(37)—(38))。
(37)
(38)
Mincher等[59]對Np(Ⅴ)的硝酸水溶液進行輻照時發現:一段時間后,溶液中產生了Np(Ⅵ)的吸收峰,這一現象可解釋為Np(Ⅴ)與水中氧化型自由基發生氧化反應。但在持續的輻照后,Np(Ⅵ)的濃度卻反而減少,即Np(Ⅵ)被重新還原成為Np(Ⅴ),這與輻射條件下Am(Ⅵ)轉化為Am(Ⅴ)的原因可能類似。Np(Ⅴ)被氧化成Np(Ⅵ)后又進一步還原的機理總結如式(39)—(46)。
(39)
(40)
(41)
(42)
(43)
(44)
(45)
(46)
在后處理PUREX流程的實際應用中,TBP萃取劑對Np(Ⅴ)的萃取性能較差,而對Np(Ⅳ)和Np(Ⅵ)選擇性高,但Np(Ⅳ)會影響Pu(Ⅳ)的價態,所以人們希望使Np更多以Np(Ⅵ)形式存在。Precek等[61]希望通過引入一些物質來滿足這一要求。通過在含Np溶液中加入V(Ⅴ),以期發生反應(47)并清除部分HNO2。
(47)
同時加入甲基脲(MU),以期清除HNO2,見圖2所示,而這有利于Np(Ⅴ)氧化成Np(Ⅵ)(式(48))。
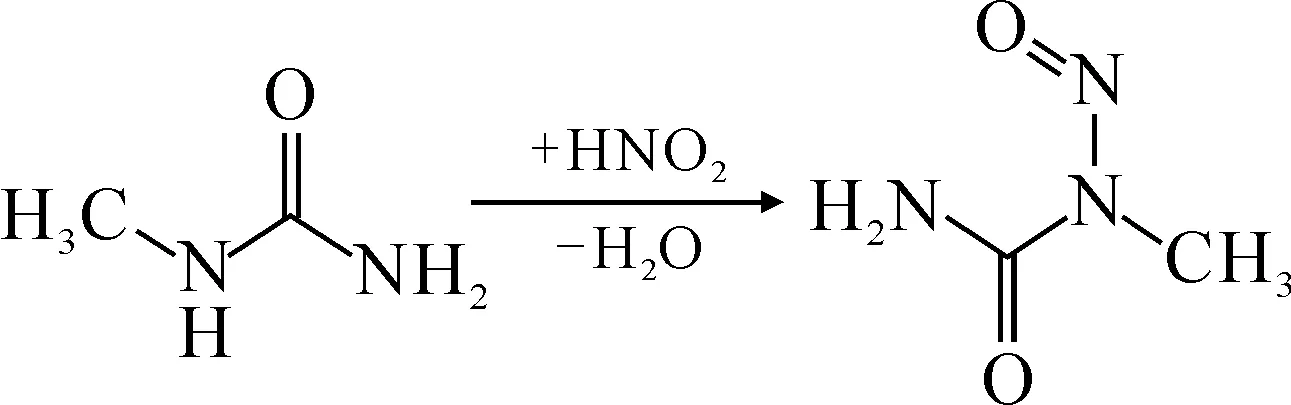
圖2 MU與HNO2的反應[61]Fig.2 Reaction between MU and HNO2[61]

(48)
Precek等[61]實驗結果發現:V對Np價態的影響不是很明顯,雖然用紫外光譜(UV光譜)檢測到了V(Ⅳ),但實驗證明V(Ⅴ)并不是被HNO2直接還原,更有可能是被輻解產物中的自由基還原。在MU的濃度達到50 mmol/L以上時,通過增加MU可以促進Np(Ⅵ)濃度的提升,但是從數據上看,提升的程度也不夠明顯。由UV光譜分析得到:在60 kGy的累積輻射劑量時,90%的Np(Ⅵ)轉化成Np(Ⅳ)的形式存在。這對Np的提取有利,但對Pu的萃取卻不利。因為Np(Ⅳ)具有還原性,會使Pu(Ⅳ)還原成為Pu(Ⅲ),PUREX流程中的萃取劑TBP對Pu(Ⅲ)的分配系數并不高。

(49)
這使得低硝酸濃度Am(Ⅲ)體系下的水溶液的G(HNO2)較小。而對于低硝酸濃度Pu體系下的水溶液而言,主要有兩點原因使得G(H2O2)較高:
(1) Pu(Ⅳ)本身會與H2O2反應(式(50))。
(50)
(2) Pu的價態變化在G(HNO2)上也起到了關鍵性的作用,Pu在水溶液中會發生反應(51)—(56)。
(51)
(52)
(53)
(54)
(55)
(56)
在Pu(Ⅳ)轉變成為Pu(Ⅲ)后,可發生反應(57)。
(57)
Pu(Ⅲ)轉化成為Pu(Ⅳ)后又會進行上述反應,使得Pu(Ⅳ)充當著催化劑的角色。這一步反應產生的·NO2會進一步促進HNO2的生成(反應(58)—(59))。
(58)
(59)
這與Liu等[20]認為Pu(Ⅳ)作為一種催化劑,能增加G(HNO2)的觀點一致,反應機理如式(60):
(60)
當濃度升高時,G(HNO2)主要還是來自于對硝酸的輻解,這時Pu價態對G(HNO2)的影響逐漸變小,以至于G(HNO2)主要來自于Pu和Am對HNO3的α輻解。
由于H2的易燃性,H2的產生在后處理流程中也是一個需要被考慮的問題。Gregson等[63]對含有Pu(Ⅳ)和Am(Ⅲ)的硝酸溶液的α自輻解產氫進行了一定的探究,發現在硝酸濃度增加時,對于含有Pu或Am的α自輻解體系,G(H2)反而下降。硝酸體系產生H2的機理如式(61)—(64)。
(61)
(62)
(63)
(64)
70%的G(H2)來自于上述反應(64)。在體系為弱酸性時,溶液中的H+會和e-的前體及e-發生反應(65)—(66):
(65)
(66)
這會促進H2的產生。但當硝酸濃度增加時,按照Horne等[24]的理論,會同時發生反應(11)、(67)—(69):
(67)
(68)
(69)
使得H2的濃度降低。而Pu(Ⅳ)和Am(Ⅲ)在弱酸體系中由于自身具有電子清除劑的作用也能起到抑制G(H2)的能力(式(70)—(71))。
(70)
(71)
但硝酸濃度較高時,Pu(Ⅳ)和Am(Ⅲ)影響G(H2)的能力將由于反應(11)的增強而相對減弱。
3 稀釋劑的輻解
由于萃取劑的粘度大,密度與水比較接近,直接用萃取劑作為有機相,容易產生第三相,降低萃取性能。為了解決這一問題,必須采用稀釋劑。
后處理中常見的稀釋劑主要以正十二烷(NDD)和煤油為主。隨著后處理的進一步研究,一批新的萃取體系被研究出來,如以正辛醇為稀釋劑的6,6′-雙-(5,6-二烷基-1,2,4-三嗪-3-基)-2,2′-聯吡啶(BTBP)和1,3-雙(4-吡啶基)丙烷(BPP)類萃取體系以及以環己酮為稀釋劑的BTBP萃取體系等。離子液體ILs具有優良的物理化學性能,比如提供用于離子結合的粒子、較低的揮發性、高電導率、高的極性和極高的金屬離子溶解度,在實驗室范圍作為有機膦類、酰胺類和含氮雜環類萃取劑的稀釋劑也已經開展了較多的研究[64-67]。但基于國內已有兩篇描述較為詳細的綜述介紹近年來離子液體的輻射效應研究[68-69],本文主要介紹近十年來已經應用的烷烴類稀釋劑的輻解效應研究。
Mincher等[70]對直鏈烷烴類的稀釋劑的輻解已經進行了較為詳細的綜述。直鏈烷烴在輻解過程主要會生成以下物質(式(72))。

(72)
在輻解過程最先發生反應(73)—(75)。
(73)
(74)
(75)
由于在直鏈烷烴的輻解過程中只有小于10%的輻解產物是來自于直接輻解,剩余部分主要通過電離過程產生[71]。上述反應是已知探測尺度下直鏈烷烴在輻照后最先誘發的反應,對直鏈烷烴的輻解有著重要的影響。但由于時間尺度較短,實驗測定較為困難。Kondoh等[72-73]首次使用飛秒級脈沖輻解裝置對NDD的輻解過程中發生的上述反應進行了探究,研究表明輻照產生的RH·+*只有7 ps的壽命。在7 ps內,73%的RH·+*會發生退激反應(反應(74)),并與電子發生重組(反應(75)),剩余的27%的RH·+*直接與電子發生重組。最終RH·+*形成自由離子、RH*、RH**等形態,并發生一系列電離反應。
4 乏燃料的氧化溶解
為了評估地下水與乏燃料潛在接觸后地質處置庫的安全性,有必要在處置多年后確定乏燃料的溶解特性。因為未“燃燒”充分的乏燃料會釋放出α粒子,在水中引起H2O2等氧化性物質濃度的累積,從而會使得固態的乏燃料的價態發生變化,轉化成為可溶于水的價態,導致在水溶液中釋放更多的α粒子,這一過程對后處理處置十分不利。研究乏燃料在水中的溶解是一個有實際意義的課題。近十年內已有較為廣泛的研究[74-76]。圖3描述了UO2溶解過程的反應機理[77]。
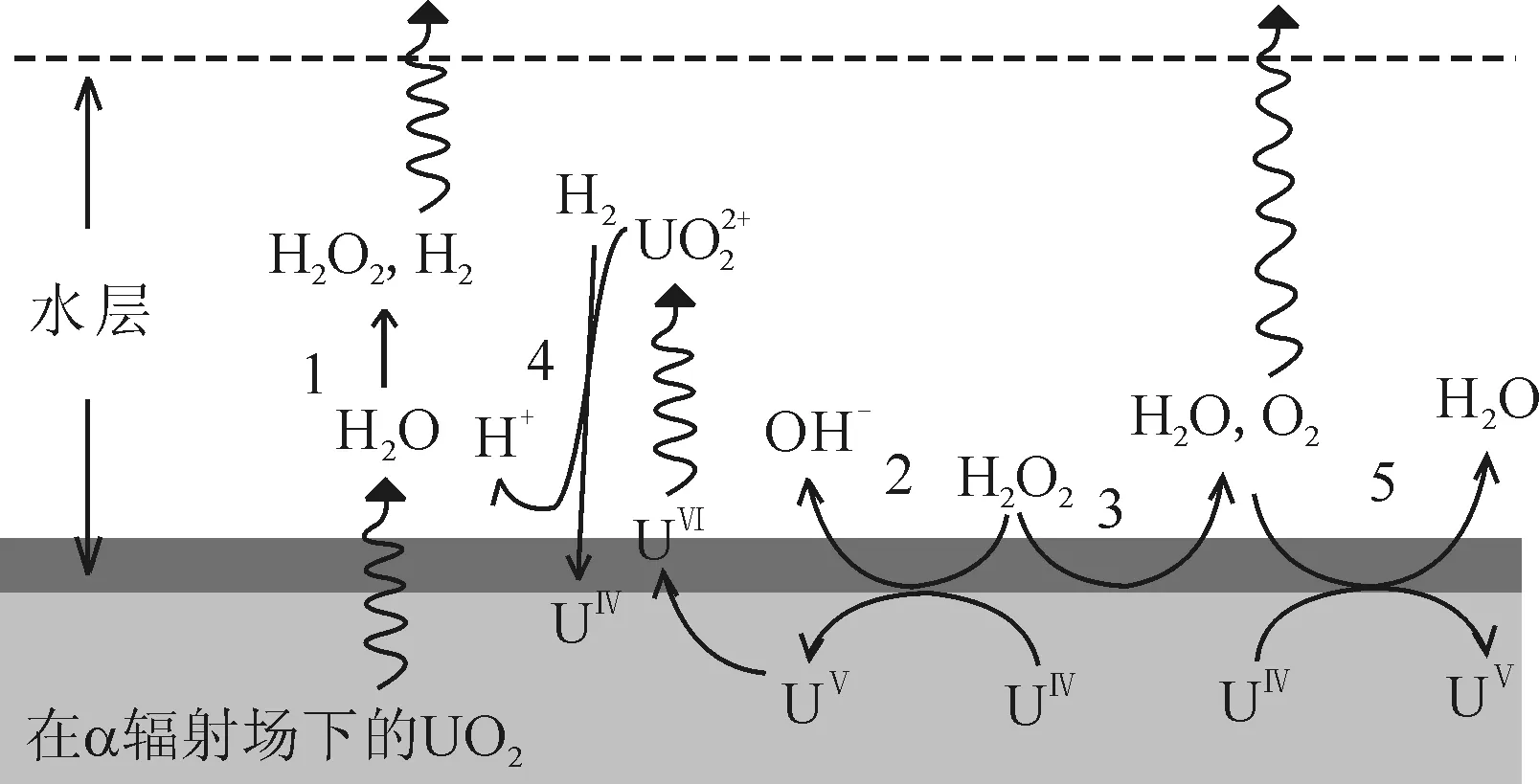
1—5——反應編號圖3 α輻照條件下UO2的溶解機理[77]Fig.3 Dissolution mechanism of UO2 under α-irradiation conditions[77]
Liu等[78]對含有放射性的UO2的封閉體系進行模擬研究,H2O2、O2和H2均會對UO2的溶解產生影響。在封閉的自輻解體系里,O2和H2會因為水的輻解產生并最終到達穩定的濃度。O2會增加溶解速率,而H2的累積則會抑制這一過程直到一個可以忽略的水平。所以UO2的溶解速率主要取決于H2O2的生成速率。
H2O2的生成途徑之一為式(76)。
(76)
Shilov等[79]認為該過程中O—O鍵的結合會釋放出能量,一部分給溶劑水分子,一部分會使得H2O2處于激發態,因而以H2O2作為反應物的化學反應更容易達到反應閾能,使得溶液中因輻解產生的H2O2比外加的H2O2與體系內的物質反應速率更快。
對后處理地質處置過程中UO2與輻解的氧化產物如H2O2的接觸已經開展廣泛研究,但至今均相NpO2和PuO2的放射性溶解動力學的實驗在公開文獻中卻基本上沒有。Pehrman等[80]探究了含有NpO2和PuO2的H2O2水溶液消耗的動力學,并將它們的行為與UO2和H2O2的行為進行了比較。實驗結果發現,在H2O2存在的條件下,錒系元素的固體氧化物的溶解速率大小分別為UO2>NpO2>PuO2。而溶液中無H2O2時,NpO2和PuO2的溶解速率分別下降90%和85%。進一步證明了H2O2在地質處置中對氧化溶解十分不利。
Odorowski等[81]進一步探究了MOX燃料在有氧和無氧的碳酸鹽水溶液中長期(一年)輻照的氧化溶解過程和溶解的錒系元素的形態。相比于PuO2,UO2會優先氧化溶解。這與Pehrman等[80]的結論保持一致:釋放的U元素主要以溶解度較高的碳酸鹽絡合物的形式存在,而Pu和Am主要富集在容器TiO2的壁面上;12%的Am以膠體的形式存在于水溶液中,而溶解在溶液中的Pu則以部分膠體和部分水合無定形Pu(Ⅳ)氧化物形式存在。
5 總結和展望
針對后處理過程中不同的處理目的已經開發出了多個流程和萃取劑,而萃取劑的輻照穩定性很大程度上影響著流程的好壞和處理效果。所以新型萃取劑的合成主要是為了實現更高的萃取效率和更優異的輻射穩定性。由于鑭系元素偏軟,基于軟原子N或S配體的萃取劑成為目前的研究趨勢,已經開發出來了二甘醇酰胺(DGAs)、雙-三嗪基-吡啶類(BTPs)等含N萃取劑和亞楓等含S萃取劑。一方面通過改性或者接枝到杯冠等結構中以提高萃取性能,另一方面增加保護基團從而使得激發態的萃取劑自由基或離子通過復合、空穴轉移或者自由基轉移等反應保護關鍵基團。由于稀釋劑的輻解自由基對萃取劑的影響也比較大,人們發現離子液體作為稀釋劑時比傳統的NDD對萃取劑的影響更小。0.001 mol/L C4DGA/[C8mim][NTf2]型萃取體系的D(Am)高達5 500,在500 kGy的累積輻照后,D(Am)只減少了7%[66]。相比于TBP/NDD等傳統萃取體系,該新型萃取體系的萃取性能和輻射穩定性有很大的提高。因此發展新型的離子液體和萃取劑是未來的趨勢。
迄今為止,對萃取劑輻射穩定性的研究絕大多數采用穩態輻照后性能表征和穩態產物分析的方法,對硝酸水溶液與鑭系元素的水溶液輻解也多采用穩態輻照后對溶液光譜進行分析的方法。然而,對輻解過程中的瞬態產物和短時間尺度內輻解基元反應動力學仍缺乏系統研究,這無疑不利于深入認識其輻解機理。因此,需要加強輻射化學的基礎反應過程研究,特別是對短壽命中間產物的研究。飛秒、皮秒級等超快脈沖輻解技術將會有利地促進對新型萃取劑等輻解機理的研究。實際上,許多化學基礎反應過程都發生在皮秒甚至亞皮秒時間范圍內,如電子、正離子和負離子的溶劑化過程,離子對的復合,液體中激發態的形成和衰減,納米粒子的成核過程,極度狹窄空間體系中的電子轉移和反應等。在乏燃料后處理中廣泛使用的烷烴類有機溶劑,介電常數低,Onsager半徑大,電離輻射作用后生成的電子很快地復合,因此直接觀測的時間在皮秒及亞皮秒范圍。被認為在新核能系統、核燃料循環中頗有前景的介質體系如超臨界流體、離子液體等,以及在熔鹽堆和乏燃料干法后處理中使用的高溫熔鹽體系等,對這些體系輻解機理的研究,也只有超快脈沖輻解技術才能勝任。
猜你喜歡
汽車實用技術(2022年15期)2022-08-19 02:48:28
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
今日農業(2020年20期)2020-12-15 15:53:19
中國外匯(2019年17期)2019-11-16 09:31:14
能源(2018年10期)2018-12-08 08:02:48
能源(2016年10期)2016-02-28 11:33:30
汽車實用技術(2015年8期)2015-12-26 09:01:02
現代企業(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
汽車與新動力(2013年3期)2013-03-11 16:08:03
