牙鲆甲狀腺激素受體TRαA介導甲狀腺激素調控的靶基因鑒定
2020-04-06 05:09:56付元帥施志儀季文瑤陶家康
水生生物學報 2020年2期
謝 燕 付元帥 施志儀 季文瑤 陶家康
(上海海洋大學農(nóng)業(yè)部淡水水產(chǎn)種質資源重點實驗室, 上海 201306)
牙鲆Paralichthys olivaceusTemminck & Schlegel屬于鰈形目, 在仔魚向稚魚的胚后發(fā)育中經(jīng)歷劇烈的變態(tài)過程, 表現(xiàn)為右眼左移、體位從側臥變?yōu)槠脚P、浮游轉為底棲生活等。甲狀腺激素(Thyroid Hormone, TH)在鰈形目魚類變態(tài)過程中起著關鍵性的調控作用[1—3], 直接控制著仔魚變態(tài)的進程。T3(3, 3′, 5-triiodo-L-thyronine,三碘甲狀腺原氨酸)是TH發(fā)揮生理作用的主要形式。TH是通過甲狀腺激素受體(Thyroid Hormone Receptors, TRs)和非甲狀腺激素受體發(fā)揮作用[4,5]。甲狀腺激素受體屬于核受體超家族中的一員, 是配體T3誘導的轉錄因子, 在介導T3的作用過程中處于核心地位, 通過招募多種共激活子或共抑制子來實現(xiàn)基因轉錄的激活或抑制。TRs分子在結構上分為6個區(qū), 從氨基端到羧基端依次為A-F區(qū), 組成3個功能域: 轉錄激活域AF-1(A/B區(qū)), DNA結合域DBD(C區(qū)), 鉸鏈區(qū)(D區(qū)), 配體結合域LBD(E區(qū))[6,7]。TRs與維甲酸X受體(Retinoid X receptors)及其他核受體形成異二聚體結合到靶基因啟動子區(qū)域的甲狀腺激素應答元件上(Thyroid Hormone Response Elements,TREs), 從而調控靶基因轉錄[8,9]。TREs通常包含兩個或更多個串聯(lián)排列的六聚體半位序列AGGT(C/A)A[10], 具有3種識別類型[11]: 回文結構(Palindrome, AGGTCATGACCT, 如生長激素Ⅰ即Gh1)、直接重復(Direct Repeat, 最常見DR4類型, AGGTCANNNNAGGTCA, 如kruppel-like因子9即Klf9)和倒置回文結構(Everted Repeat, 最常見6 bp間隔,TGACCTNNNNNNAGGTCA, 如髓磷脂堿性蛋白即myelin basic protein, Mbp)。牙鲆含有TRαA、TRαB和TRβ三種受體, TR亞型(TRαA、TRαB和TRβ1、TRβ2)在牙鲆變態(tài)期間基因表達具有時間特異性和組織特異性:TRαA在變態(tài)高峰期急劇增加,高峰期后又迅速下降;TRαB在仔魚發(fā)育整個過程中都很低;TRβs的表達水平在變態(tài)高峰增加, 于變態(tài)高峰后達到峰值, 高水平的TRβs在變態(tài)完成的稚魚中尚在持續(xù)[3,12,13]。
那么TH是如何調控牙鲆仔魚的變態(tài)? 鑒定出TRs直接調控的靶基因是一個關鍵。通過牙鲆轉錄組及基因組TRE數(shù)據(jù)庫篩選, 結合生物信息學分析,初步確定候選靶基因atoh8(Gene ID:109627718)。atoh8(Atonal homolog 8, atonal bHLH transcription factor 8), 屬于堿性螺旋-環(huán)-螺旋(basic Helix-Loop-Helix, bHLH)轉錄因子, 參與斑馬魚視網(wǎng)膜細胞和體節(jié)肌肉細胞的發(fā)育分化, 在視網(wǎng)膜和體節(jié)發(fā)育的調控過程中,atoh8起著不可或缺的重要作用[14]。關于atoh8與甲狀腺激素調控通路的關系尚未見報道。
本研究克隆了牙鲆TRαA基因及atoh8基因5′-側翼序列各缺失片段, 并成功構建p3×Flag-TRαA和pGL3-Pro-atoh8-1517/1333/708(含候選靶基因5′-側翼序列的缺失片段), 通過在HEK293T細胞中共轉染p3×Flag-TRαA和pGL3-Pro-atoh8-1517/1333/708的雙熒光素酶報告實驗, 分別探究不同的5′-側翼序列長度和對同一5′-側翼序列進行不同處理(TRαA和T3都不加、只加TRαA或同時加TRαA與T3)對啟動子轉錄活性的影響, 以期鑒定出TRαA受體是否結合在atoh8基因5′調控區(qū)特異的TRE序列來調控該基因的轉錄并判別具體是哪個TRE位點起到了關鍵作用, 最終鑒定出atoh8是否為TRαA介導甲狀腺激素調控的直接靶基因。
1 材料與方法
1.1 材料與試劑
出膜后20d (20dph,days post hatching)、28d(28dph)的牙鲆仔魚, 采自中國水產(chǎn)科學研究院北戴河中心實驗站; HEK293T細胞購自ATCC細胞庫。DMEM、胎牛血清、青霉素-鏈霉素、0.25%胰酶-EDTA購自Gibco; 細胞裂解液、蛋白酶抑制劑PMSF、ECL化學發(fā)光試劑盒購自上海威奧; 一抗Anti-Flag Tag (1E6) Monoclonal Antibody (GNI4110-FG)購自上海睿星, 二抗HRP-conjugated Goat Anti-Mouse IgG (D110087)、5×蛋白質加樣緩沖液、平端 DNA 片段添dA試劑盒購自上海生工, 引物合成由上海生工完成; BCA蛋白定量試劑盒購自康為世紀, Anti-DYKDDDDK G1 Affinity Resin和DYKDDDDK Peptide購自金斯瑞; DNaseI、反轉錄試劑盒、FuGENE?HD轉染試劑均購于Promega; 雙熒光素酶報告基因檢測試劑盒購自YEASEN,EcoRⅠ、KpnⅠ-HF、XhoⅠ限制酶、T4 DNA Ligase購自NEB; X-gal、IPTG購自天根, 2 × Phanta Max Master Mix購自Vazyme, pEASY?-T5 Zero購自全式金,DH5α購自上海唯地; TaKaRa LATaq、pMD19-T、TB GreenTMPremix ExTaqTMⅡ (Tli RNaseH Plus)購自TaKaRa, 瓊脂糖凝膠DNA回收試劑盒、質粒小提、無內毒素質粒大提試劑盒購自Omega。p3×Flag、pGL3-Basic載體由上海海洋大學李名友教授惠贈。
1.2 方法
總RNA提取和反轉錄將28dph仔魚按照Trizol?Reagent(Invitrogen)試劑盒說明書提取總RNA。檢測總RNA純度、濃度和完整性[15], 均符合要求后置于-80℃保存; 將上述RNA用DNaseⅠ處理后, 按Su等[16]方法反轉錄, 產(chǎn)物cDNA用于PCR擴增或-20℃保存。
牙鲆TRαA基因CDS區(qū)的克隆從NCBI(https://www.ncbi.nlm.nih.gov/)數(shù)據(jù)庫中獲取牙鲆TRαA基因序列(Gene ID: 109641414), 根據(jù)其序列和p3×Flag多克隆位點設計一對引物TRαA1252-F/R(表 1), 以上述cDNA為模板, 通過2×Phanta Max Master Mix擴增CDS區(qū)全長。
擴增產(chǎn)物經(jīng)1%瓊脂糖凝膠電泳, 膠回收純化目的片段, 加A, 連接至pEASY?-T5 Zero, 轉化DH5α。挑取陽性單克隆, 經(jīng)菌液PCR驗證有大小正確的片段插入后測序, 交由上海生工完成。
重組真核表達載體p3×Flag-TRαA的構建將上述測序正確的菌液和p3×Flag擴大培養(yǎng)提質粒;用EcoRⅠ、KpnⅠ37℃雙酶切; 將鑒定正確的酶切PCR產(chǎn)物與p3×Flag載體分別進行膠回收, 產(chǎn)物按摩爾比3∶1經(jīng)T4 DNA Ligase 16℃連接16h; 轉化DH5α, 挑取陽性單克隆, 搖菌提質粒, 雙酶切驗證,再送上海生工測序驗證; 將鑒定正確的p3×Flag-TRαA擴大培養(yǎng), 提取不含內毒素的重組質粒。
HEK293T細胞培養(yǎng)及瞬時轉染HEK293T細胞培養(yǎng)于10% FBS-DMEM 培養(yǎng)液中, 置于37℃,5% CO2的培養(yǎng)箱中常規(guī)培養(yǎng), 轉染前一天接種于6孔細胞培養(yǎng)板1.5 mL不含抗生素的10% FBSDMEM中, 轉染時細胞密度為90%—95%。配制轉染復合物[3 μg p3×Flag-TRαA(實驗組)或p3×Flag(陰性對照), 7.5 μL FuGENE?HD轉染試劑, 無抗生素、無血清DMEM補至500 μL], 混勻后室溫孵育10—15min, 加到6孔板相應孔中, 未轉染質粒作為空白對照, 每組(未轉、空載、重組質粒)設4個重復, 一組用于總蛋白提取, 其余3組用于總RNA提取。培養(yǎng)6h左右換完全培養(yǎng)基, 48h后進行總蛋白和總RNA提取。
TRαA基因的RT-PCR及實時定量PCR檢測將上文中RNA反轉錄成cDNA, PCR擴增TRαA基因CDS區(qū)全長與18S定量內參基因。在CFX96 TouchTMReal-Time PCR Detection System (Bio-Rad, USA)上分別制作目的基因TRαA和內參基因18S的標準曲線, 在符合要求后, 隨后定量分析所有樣品。反應體系(20 μL): 1 μL cDNA, 各1 μLTRαA-qF/R或18S-qF/R (表 1), 10 μL TB GreenTMPremix ExTaqTMⅡ(Tli RNaseH Plus)和7.0 μL DEPC水; 反應條件:95℃ 3min; 95℃ 10s, 60℃ 30s, 采集熒光40次。該實驗中, 生物學重復n=3, 技術重復2次。
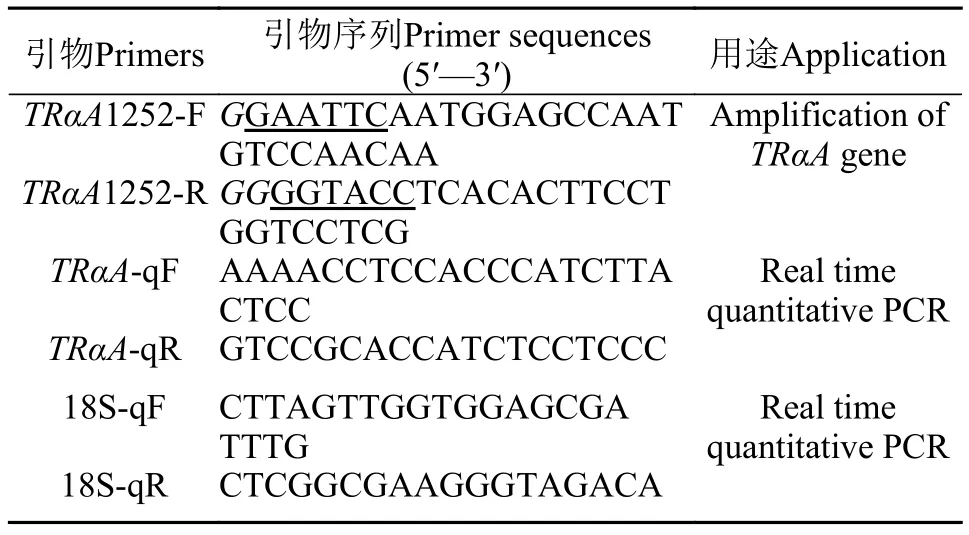
表 1 TRαA基因的擴增和定量引物Tab. 1 The primer used for cloning and real time quantitative PCR of TRαA gene
TRαA的mRNA相對表達量通過2-ΔΔCt[17]方法計算, 其結果以平均值±標準誤差(Mean±SE)來表示(n=3)。通過SigmaPlot 12.0軟件作圖分析,利用SPSS 22軟件中的One-Way ANOVA方差分析法分析不同樣品間的相對表達差異, *P<0.05時表示差異顯著。
Western blot檢測融合蛋白的表達按上文方法轉染HEK293T細胞, 經(jīng)PBS洗2次, 1000 r/min離心5min分類收集HEK293T細胞; 加入預冷含PMSF (裂解液∶PMSF=99∶1)的細胞裂解液, 現(xiàn)配現(xiàn)用, 混勻后冰上輕微搖晃10min; 4℃、13000 r/min離心5min, 取上清, 分裝使用或-80℃凍存。參考說明書用BCA試劑盒進行蛋白濃度測定。
分別吸取上述蛋白樣品10 μg, 裂解液補至20 μL,加入5×SDS-PAGE加樣緩沖液5 μL 100℃干式恒溫10min, -20℃保存或加樣進行SDS-PAGE電泳, 將分離的蛋白條帶電轉印至 PVDF 膜上, TBST洗2次(每次10min, 下述一樣); 再將 PVDF 膜浸于含5%脫脂奶粉的TBS中室溫封閉1h, TBST洗2次; 按1∶1000用TBS稀釋一抗(小鼠抗Flag標簽單抗)4℃孵育過夜, TBST洗3次; 按1∶5000用TBST稀釋二抗(HRP標記的山羊抗小鼠 IgG)37℃孵育1h, TBST洗3次; 將PVDF膜與ECL發(fā)光液避光充分接觸1min, 在暗室X光片下曝光拍照。
融合蛋白3×Flag-TRαA的純化采用T25瓶整瓶細胞轉染, 且質粒與轉染試劑比為4 μg∶8 μL,按上述方法收集細胞提取總蛋白。目的蛋白TRαA的N端融合有3×Flag標簽, 通過G1親和層析柱純化后, 分別保留總蛋白原液、流出液、洗雜液和洗脫液, 洗脫液經(jīng)無菌濾器過濾除菌后, -80℃保存,Western blot檢測純化效果。
TRαA直接調控候選靶基因的篩選與重組報告基因表達載體pGL3-Pro-atoh8的構建 通過轉錄組數(shù)據(jù)(正常樣品, 甲狀腺素處理樣品)篩查, 及基于現(xiàn)有上傳的牙鲆基因組建立的TRE數(shù)據(jù)庫篩選, 結合TREs識別類型, 從NCBI中獲取牙鲆候選靶啟動子5′-側翼序列, 利用在線網(wǎng)站http://www.fruitfly.org/seq_tools/promoter.html, http://www.cbs.dtu.dk/services/Promoter/, https://www-bimas.cit.nih.gov/molbio/proscan/, http://biogrid-lasagna.engr.uconn.edu/lasagna_search/等預測靶啟動子轉錄活性及轉錄因子結合位點。初步確定候選靶啟動子, 如atoh8(Gene ID:109627718)。
采用苯酚抽提法從20dph仔魚中提取基因組DNA。根據(jù)atoh8基因5′-側翼序列和pGL3-Basic多克隆位點設計3對引物(表 2), 固定下游引物, 改變上游引物以缺失預測出的TRE位點, 以基因組DNA為模板, 經(jīng)TaKaRa LATaq擴增先得到atoh8啟動子最長的缺失片段。
按上述方法(產(chǎn)物連至pMD19-T, 用KpnⅠ、XhoⅠ雙酶切, 酶切PCR產(chǎn)物和pGL3載體4℃連接過夜)獲得pGL3-Pro-atoh8-1517。再以pGL3-Proatoh8-1517為模板, 按如上方法獲得pGL3-Proatoh8-1333/708。
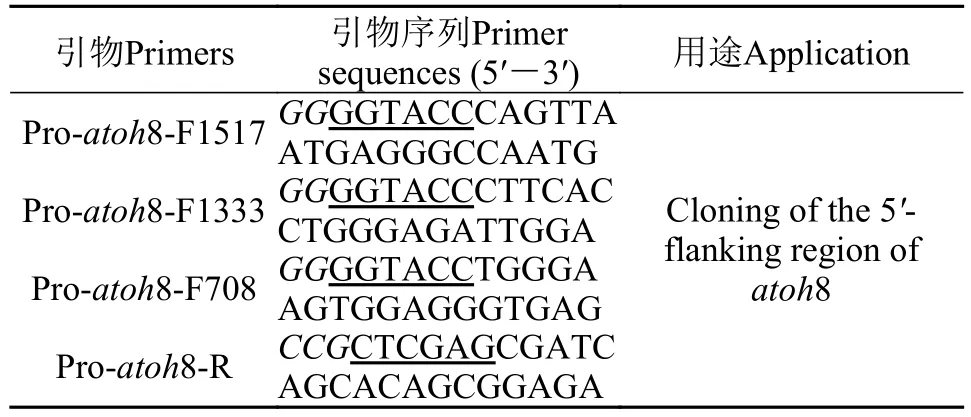
表 2 候選靶啟動子各缺失片段的擴增引物Tab. 2 The primer used for cloning each missing fragment of the candidate target promoter
雙熒光素酶報告實驗參考前文方法將HEK293T細胞接種于96孔細胞培養(yǎng)板, 配制轉染復合物[10 ng內參質粒 phRL-TK, 100 ng空載pGL3-Basic或100 ng重組質粒pGL3-Pro-atoh8-1517/1333/708(或含100 ng重組質粒p3×Flag-TRαA),0.22 μL FuGENE?HD 轉染試劑, 無抗生素、無血清DMEM補至10 μL], 混勻后室溫孵育10—15min,加到96孔細胞培養(yǎng)板相應孔中, 繼續(xù)培養(yǎng), 6h后換完全培養(yǎng)基, 再 24h后向轉染的細胞中加入0.1 μL T3(終濃度為150 ng/μL), 再24h后檢測并計算螢火蟲熒光素酶與海腎熒光素酶的比值, 每個轉染組設置3個復孔, 重復3次。數(shù)據(jù)采用SigmaPlot 12.0作圖, 并通過SPSS 22軟件中的單因素方差分析“一般線性模型”-“單變量”和LSD進行相對表達分析, 當*P<0.05時接受差異顯著。
2 結果
2.1 牙鲆TRαA基因CDS區(qū)的克隆和p3×Flag-TRαA的構建
以28dph牙鲆仔魚 cDNA 為模板進行PCR擴增,電泳結果顯示片段大小約為1.2 kb, 與NCBI上傳(XM_020105869.1)的大小相一致; 實際擴增得到片段大小是1252 bp, 并成功構建了p3×Flag-TRαA重組質粒。測序結果顯示CDS區(qū)第850個核苷酸穩(wěn)定由A突變?yōu)镚, 導致氨基酸由Ser轉變?yōu)镚ly; 920—1030 bp有兩小段, 分別是連續(xù)9 bp、連續(xù)11 bp與NCBI上傳序列始終不同, 且不同的高保真酶擴增測序結果相同。
2.2 牙鲆TRαA基因推導氨基酸序列分析及蛋白結構預測
利用ProtParam tool (https://web.expasy.org/protparam/)平臺, 分析TRαA基因編碼序列所推導蛋白質的基本理化性質, 預測蛋白質的相對分子量為47 kD, 理論等電點是7.06。Leu、Lys、Glu、Ser含量比較高, 分別為 8.4%、8.2%、7.9%、7.5%。利用SWISS-MODEL (https://www.swissmodel.expasy.org/)在線軟件, 分析TRαA的三級結構(圖 1), 結果顯示該蛋白含有8個不同長度的α 螺旋結構、1個典型的β轉角和無規(guī)卷曲。
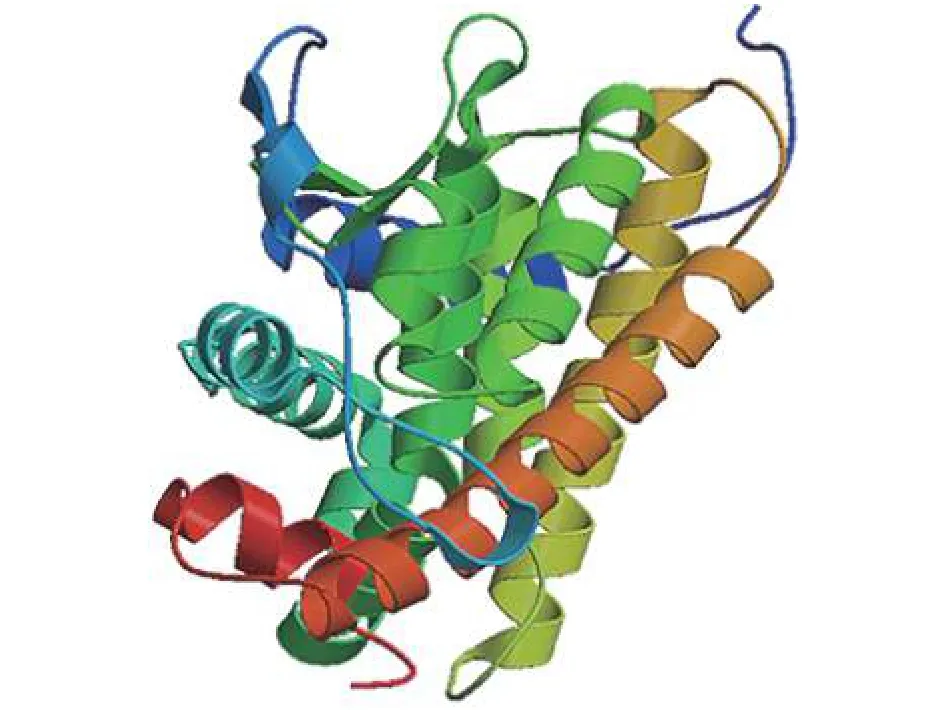
圖 1 牙鲆TRαA受體蛋白結構預測Fig. 1 Prediction of protein structure of P. olivaceus TRαA receptor
2.3 TRαA基因的RT-PCR及實時定量PCR檢測
RT-PCR產(chǎn)物電泳結果顯示, 實驗組在長度1200 bp處顯示有一特異性條帶, 與預期大小相同,而對照組均出現(xiàn)一個相應的亮度減弱且二者亮度相同的條帶, 原因可能是HEK293T細胞中存在牙鲆TRαA基因的同源序列; 實時定量PCR也顯示, 與對照組相比, 實驗組TRαAmRNA表達水平顯著升高,這都表明轉染p3×Flag-TRαA的HEK293T細胞中, 成功轉錄出牙鲆TRαAmRNA(圖 2)。
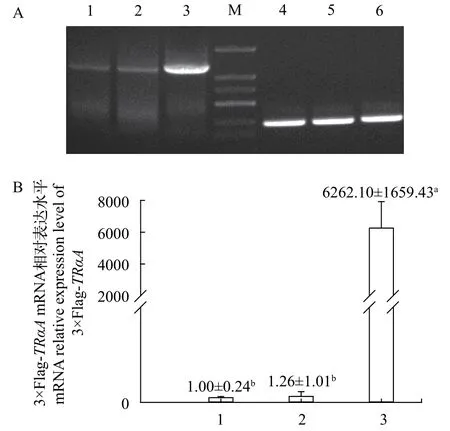
圖 2 重組質粒或空載轉染HEK293T細胞TRαA mRNA RTPCR及實時定量RT-PCR表達分析Fig. 2 RT-PCR and real time quantitative RT-PCR analysis of P.olivaceus TRαA mRNA in the HEK293T transfected with recombinant or vacant vector using 18S as an internal reference
2.4 融合蛋白3×Flag-TRαA的Western blot檢測
BCA法(圖 3)測得未轉染、空載轉染和重組質粒轉染HEK293T細胞提取的總蛋白濃度分別是2.055、1.650和3.440 μg/μL, SDS-PAGE上樣10 μg分離蛋白條帶后, Western blot分析顯示重組質粒轉染組檢測到一條50 kD大小條帶, 其蛋白分子量與3×Flag-TRαA融合蛋白的大小相符; 而未轉染及空載轉染組未檢測到目的條帶(圖 4)。原則上空載轉染組可被抗Flag標簽單抗特異性識別, 只因3×Flag蛋白分子與目標融合蛋白相比太小僅2.9 kD,分離膠底部被切除或已跑脫導致無法檢測到。
2.5 目的蛋白的分離純化
通過Western blot在洗脫液中可檢測出50 kD左右的蛋白, 即通過親和層析、過濾除菌得到了純化的融合蛋白3×Flag-TrαA(圖 5)。
2.6 候選靶啟動子的篩選與重組報告載體構建
根據(jù)atoh8基因啟動子轉錄因子結合位點預測結果, 分別設計了3個上游引物, 1個下游引物, 試以牙鲆基因組DNA 為模板擴增atoh8基因5′-側翼序列的各缺失片段, 電泳結果顯示其大小約為1500、1300和700 bp, 實際擴增出的片段大小為1517、1333和708 bp (圖 6A為其實際擴增測序的序列, 引物與預測TREs位點已標出), 與NCBI上傳的大小稍有出入, 并成功構建了重組質粒pGL3-Pro-atoh8-1517/1333/708。
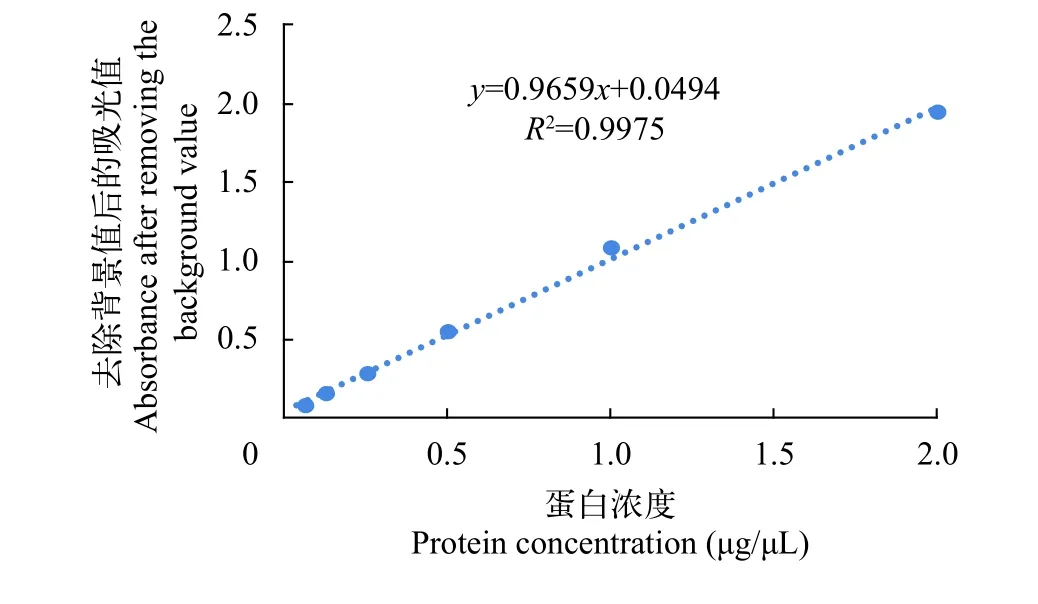
圖 3 BCA法測定蛋白濃度標準曲線Fig. 3 Determination of protein concentration standard curve by BCA method
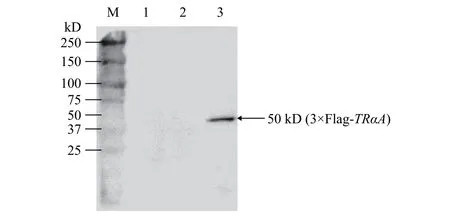
圖 4 融合蛋白3×Flag-TRαA的Western blot分析Fig. 4 Western blot analysis of fusion protein 3×Flag-TrαA

圖 5 純化融合蛋白3×Flag-TRαA的Western blot分析Fig. 5 Western blot analysis of purified fusion protein 3×Flag-TRαA
2.7 atoh8基因啟動子研究
根據(jù)預測TRE位點所在位置, 分別截取了3個缺失片段, 大小分別為1517、1333和708 bp, 依次包含3個、2個、1個TRE位點, 測序結果如圖 6A所示,各缺失片段起止區(qū)段如圖 6B所示。分別檢測每一缺失片段的啟動子轉錄活性, 對每一缺失片段進行3種處理(不加TRαA和T3、只加TRαA或同時加TRαA與T3)后再分別檢測其對啟動子轉錄活性的影響, 結果如下: 圖 7A顯示, 與pGL3-Basic轉染組(陰性對照)相比, pGL3-Pro-atoh8-1517/1333/708轉染組報告基因均表達, 表明Pro-atoh8-1517/1333/708都具有啟動子轉錄活性, 且Pro-atoh8-1333活性最高, Pro-atoh8-1517活性最低; 圖 7D顯示, 與僅轉染pGL3-Pro-atoh8-708相比, 同時轉染pGL3-Proatoh8-708和p3×Flag-TRαA的報告基因的表達量無顯著變化, 再加T3后也無顯著變化, 說明此缺失片段不存在TRαA受體介導T3調控的TRE位點; 圖 7C則表明, 與只轉染pGL3-Pro-atoh8-1313相比, 同時轉染pGL3-Pro-atoh8-1313和p3×Flag-TRαA的報告基因的表達量顯著降低, 且這種降低在加T3后其又顯著上調, 說明在Pro-atoh8-1333中存在1個TRE位點; 圖 7B顯示, 與只轉染pGL3-Pro-atoh8-1517相比,同時轉染pGL3-Pro-atoh8-1517和p3×Flag-TRαA的報告基因的表達量略有升高, 再加T3后其顯著升高, 表明Pro-atoh8-1517存在2個TRE位點(包括Proatoh8-1313的1個TRE位點); 顯然, 無論是Pro-atoh8-1517還是Pro-atoh8-1333, T3加入后都會誘導報告基因的轉錄水平顯著升高, 即啟動子轉錄活性顯著增強, 因此說明, T3通過結合在atoh8基因啟動子區(qū)(-1497— -688)特異的2個TRE序列上的TRαA受體來調控該基因的轉錄。
3 討論
為了鑒定出TH-TRαA信號通路上直接調控的靶基因, 綜合預測出候選靶基因atoh8(Gene ID:109627718), 克隆其5′-側翼序列各缺失片段并成功構建pGL3-Pro-atoh8-1517/1333/708(分別含TRE位點: 3、2、1)。本研究克隆了牙鲆TRαA基因完整CDS區(qū), 全長1251 bp (5′端多加1 bp保證TRαA受體正確翻譯), 且TRαA受體DNA結合結構域(DBD)非常保守, 為調控下游靶基因的轉錄激活與抑制所必需。已有研究[7,18]證實, DBD功能域最保守, 內有2個鋅指結構, 第一鋅指內含有P盒, 為識別并結合TREs之AGGTCA(TRs半位)所必需; 第二鋅指內含有D盒, 識別DR4元件(兩半位之間間隔4個核苷酸)兩半位(half-site)的間隔并參與受體二聚體的形成。RT-PCR、實時定量PCR和Western blot分析均表明成功構建p3×Flag-TRαA重組真核表達載體, 在目的蛋白N端融合Flag純化標簽, 由于3×Flag標簽分子量相對較小, 因此幾乎不會影響蛋白質的活性,純化產(chǎn)物可直接用于蛋白功能的研究。3×Flag-TRαA的外源性真核表達特異性強, 具有翻譯后加工修飾功能, 使其結構、糖基化方式更接近天然蛋白且生物活性穩(wěn)定[19]。

圖 6 報告基因質粒pGL3-Pro-atoh8-1517/1333/708示意圖Fig. 6 Schematic diagram of the reporter gene plasmid pGL3-Pro-atoh8-1517/1333/708
進而為了驗證TRαA受體與atoh8基因5′-側翼序列是否存在相互作用, 針對上述截取的atoh8基因5′-側翼序列的3個缺失片段(大小分別為1517、1333和708 bp, 依次包含3個、2個、1個TRE位點),及相應成功構建的重組質粒pGL3-Pro-atoh8-1517/1333/708, 采用雙熒光素酶報告實驗, 首先檢測了atoh8基因3個不同長度(1517、1333和708 bp)的5′-側翼序列對其啟動子轉錄活性的影響, 其次針對每一缺失片段, 進行了3種處理: 不作任何處理(TRαA和T3都不加)、只加TRαA或同時加TRαA與T3, 分別檢測其對啟動子轉錄活性的影響。需要指出的是, 截取的atoh8基因5′-側翼序列長度為708 bp時, 相關轉染組結果顯示TRαA受體和T3對其啟動子活性并無顯著作用, 故不進行分析討論, 以下僅討論關于pGL3-Pro-atoh8-1517/1333轉染組的情況。研究表明, 與對照組相比, 上述2個缺失片段都具有啟動子轉錄活性, 其中截取atoh8基因5′-側翼序列長度為1333 bp時的活性最高, 是長度為1517 bp時的大約3倍。據(jù)報道, 并非是截取基因的5′-側翼序列越長其啟動子轉錄活性越高[20], 本文結果亦如此。與僅轉染pGL3-Pro-atoh8-1313的實驗組相比,同時轉染pGL3-Pro-atoh8-1313和p3×Flag-TRαA的報告基因的表達量顯著降低, 但這種降低可被T3顯著上調, 表明此缺失片段存在TRαA介導T3調控的TRE位點。對于pGL3-Pro-atoh8-1517轉染組, 雖然pGL3-Pro-atoh8-1517包含pGL3-Pro-atoh8-1333的TRE位點, 但在TRαA受體加入后, 與不作任何處理組相比, 其啟動子轉錄活性反而略有升高, T3加入后又使得其啟動子轉錄活性顯著升高, 這提示在加入與pGL3-Pro-atoh8-1333轉染組等量的TRαA受體后, pGL3-Pro-atoh8-1517 5′端第1個TRE位點與第2個TRE位點結合TRαA受體, 綜合效應是第1個TRE位點中和了第2個TRE位點TRαA受體對靶基因轉錄水平的降低, 說明第1個TRE位點起了一個正向調節(jié)作用。也就是說, pGL3-Pro-atoh8-1517包含的5′端起第1個TRE位點與第2個TRE位點通過TRαA受體介導T3調控著atoh8基因的表達水平, 避免表達過強或不足, 這可能是機體實現(xiàn)精準調控atoh8基因表達的一種方式。但無論是pGL3-Proatoh8-1517轉染組還是pGL3-Pro-atoh8-1333轉染組, T3對atoh8的轉錄水平都是一種正向調節(jié)作用。
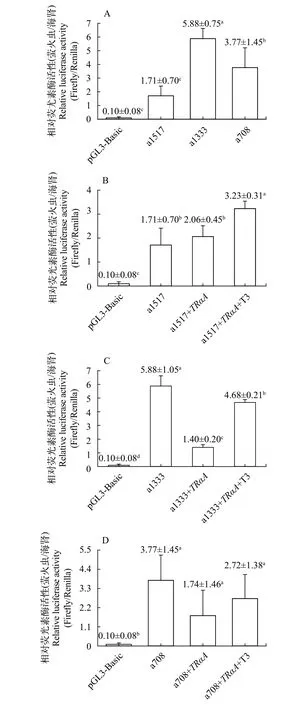
圖 7 牙鲆atoh8基因啟動子活性測定Fig. 7 Determination of promoter activity of P. olivaceus atoh8 gene
綜上所述, 本研究結果表明pGL3-Pro-atoh8-1517包含2個TRE位點, 且TRαA受體通過DNA結合結構域(DBD)結合在atoh8基因5′調控區(qū)-1497—-688特異的TRE序列來調節(jié)該基因的轉錄, 由此證明atoh8是TRαA介導TH調控的直接靶基因。本研究為深入探究甲狀腺激素受體TRαA介導甲狀腺激素調控的信號通路提供了基礎依據(jù)。
