高效液相色譜-質譜法測定牛Ⅰ型膠原含量
2020-05-09 08:13:18邢芳毓高建萍張貴鋒王明林
分析測試學報 2020年4期
關鍵詞:特征
邢芳毓,高建萍,張貴鋒*,王明林*
(1.山東農業大學 食品科學與工程學院,山東 泰安 271018;2.中國科學院過程工程研究所 生化工程國家重點實驗室,北京 100190)
膠原是哺乳動物體內最豐富的蛋白質,對于維持組織的機械穩定性具有重要作用[1-3]。目前已發現的膠原有29種[4],均為3條肽鏈組成的右旋三螺旋結構,且三螺旋結構區域內的肽鏈由規則重復的每2個氨基酸出現1個甘氨酸的序列組成。膠原作為細胞外基質的主要成分,其相互交聯成網狀結構并與其它蛋白及多糖類結合,以給予細胞和組織一定的機械強度和支撐。膠原具有低免疫原性、生物相容性、止血性和可生物降解性等生物性能,使其在生物醫學中成為最常用的材料之一,如藥物輸送系統、傷口敷料、皮膚、血管等產品[5-8]。因此,在膠原相關的研究中,膠原的定量分析成為產品研發及質量控制的關鍵。
目前膠原的定量方法眾多,如毛細管電泳法[9]、比色法[10]、高效液相色譜法[11]和免疫學檢測法[12]等,但在定量的準確性等方面均存在一定局限性。例如,基于羥脯氨酸的定量方法,只能測得總膠原含量,無法區分不同類型膠原;酶聯免疫吸附測定法要求樣品為中性溶液,但大部分膠原為酸溶性,中性條件下幾乎不溶,極大地影響檢測的準確度;利用免疫熒光染色定量組織中的膠原,需進行組織切片,而Ⅰ型和Ⅲ型膠原在皮膚真皮層中分布不均勻[13],另外該方法利用圖像軟件分析染色結果,準確度較低,誤差較大。
離子阱質譜具有多級質譜功能,可以分析化合物結構,對多肽的序列進行識別[14]。不同物種、不同類型膠原的氨基酸序列彼此不同,可用液相色譜-質譜聯用技術識別出不同類型膠原中的特征多肽,從而實現膠原的類型識別[15]。三重四極桿質譜同樣可用于結構分析,其比離子阱質譜靈敏,更適用于微量物質的定量分析[16]。胰蛋白酶降解過程中膠原的斷裂具有一定規律性(胰蛋白酶的酶切位點在精氨酸或賴氨酸的羧端),在相同的酶解條件下,可根據特征肽段的序列進行膠原定量。本文基于高效液相色譜-質譜(HPLC-MS)聯用技術,在比較不同類型的牛膠原氨基酸序列的基礎上,篩選出牛Ⅰ型膠原的特征肽段,并利用特征肽段對市售膠原海綿樣品中的牛Ⅰ型膠原進行定量,為膠原的準確定量提供了方法依據。
1 實驗部分
1.1 儀器與裝置
液相色譜-離子阱質譜由1100液相色譜(美國Agilent公司)和LCQ DecaXP電噴霧質譜(美國Thermo Fisher公司)組成;高效液相色譜-三重四極桿質譜由U3000液相色譜、TSQ Quantum ACCESS MAX質譜(美國Thermo Fisher公司)組成;數據采集與處理軟件為Xcalibur1.3。
1.2 材料與試劑
牛Ⅰ型膠原海綿(河北考力森生物科技有限公司);胰蛋白酶(序列純,豬源,Promega公司);乙腈、甲醇(色譜純,Merck公司);甲酸(色譜純,Aladdin公司);GFSGLDGAK特征肽段標準品(純度為99.28%,上海強耀生物科技有限公司);碳酸氫銨(NH4HCO3,分析純,西隴科學股份有限公司)。
1.3 樣品前處理條件
取膠原海綿樣品10 mg加入10 mL NH4HCO3緩沖液(0.05 mol/L),于沸水浴中加熱20 min,待冷卻至常溫后,加入胰蛋白酶(200 μg),37 ℃酶解16 h,再于沸水浴中加熱5 min使胰酶滅活,離心(10 000 r/min)10 min,待上樣。
1.4 凝膠過濾分析條件
色譜條件參考文獻方法[17]。
1.5 液相色譜-離子阱質譜聯用分析條件
1.5.1 色譜條件色譜柱:Zorbax SB-C18(150 mm×2.1 mm,5 μm);流動相:A為水(0.1%甲酸),B為乙腈(0.1%甲酸);洗脫梯度:0~5 min,5% B;5~40 min,5%~40% B;40~80 min,40%~75% B;流速:0.2 mL/min;進樣量:10 μL。
1.5.2 質譜條件ESI電離;噴霧電壓:4.5 kV;鞘氣流速:約400 kPa(60 arb);掃描范圍(m/z):300~2 000;正離子監測模式;精確質量數掃描(Zoom scan)和二級質譜掃描(MS/MS)均為數據依賴性掃描;二級質譜的碰撞能量為35%。
1.6 液相色譜-三重四極桿質譜聯用分析條件
1.6.1 色譜條件色譜柱:Zorbax SB-C18(150 mm×2.1 mm,5 μm);流動相:A為水(0.1%甲酸);B為60%乙腈+40%水(0.1%甲酸);洗脫梯度:0~25 min,5%~50% B;25~26 min,50%~90% B;26~35 min,90% B;35~36 min,90%~5% B;36~45 min,5% B;流速:0.2 mL/min;進樣量:10 μL。
1.6.2 質譜條件離子源:電噴霧離子源;掃描方式:正離子掃描;檢測方式:選擇離子監測(m/z851.4);噴霧電壓:3.5 kV;鞘氣壓力:241.3 kPa;輔助氣壓力:約1.2 kPa(8 arb);鞘氣輔助氣壓力:0.6 MPa;離子傳輸管溫度:270 ℃;離子源蒸發溫度:100 ℃。
1.7 水分與熾灼殘渣測定
按照《中華人民共和國藥典》[18]中0832和0841規定的方法進行。
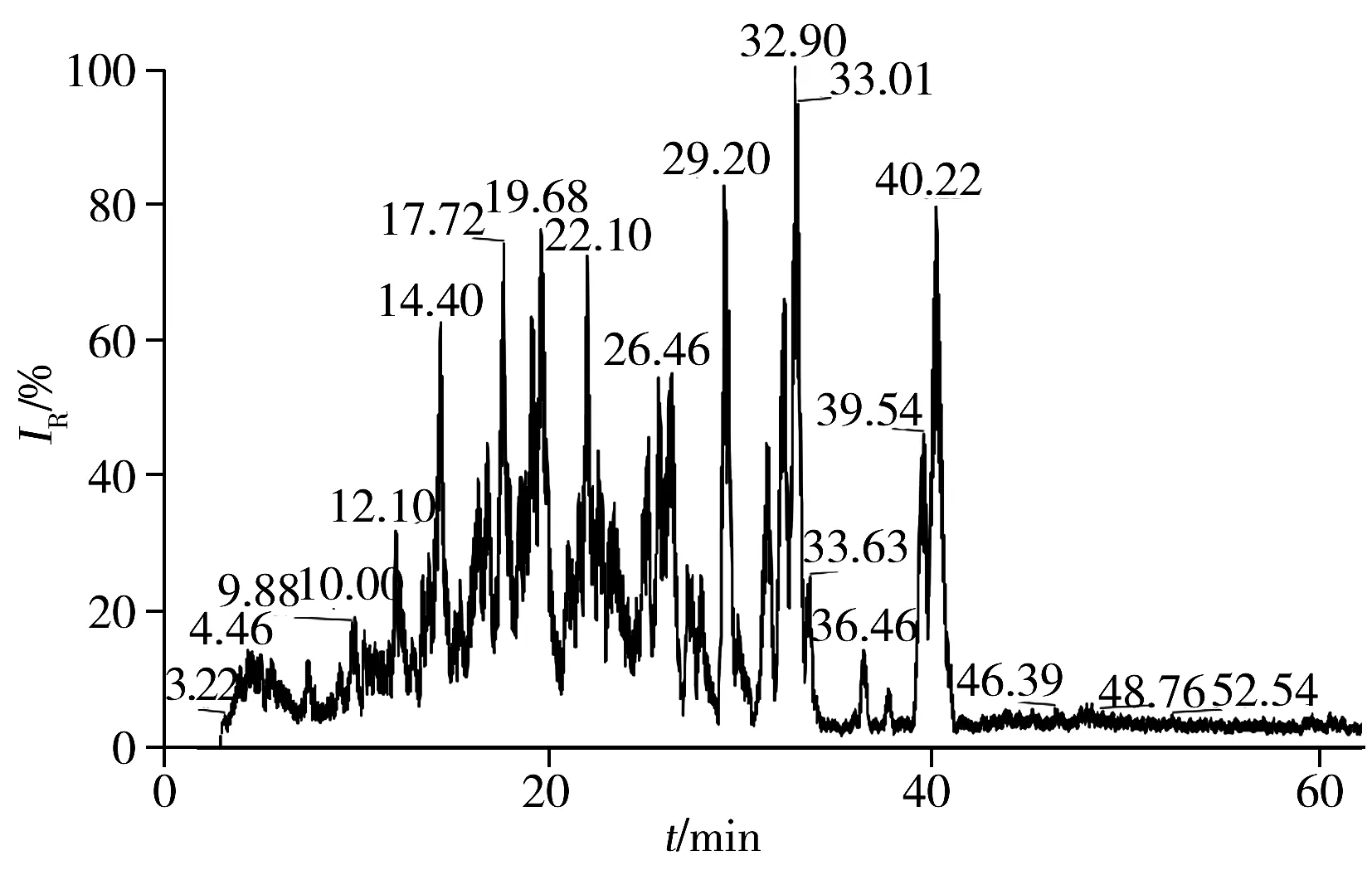
圖1 膠原海綿酶解產物的總離子流圖Fig.1 Total ion current chromatogram of digested collagen sponge
2 結果與討論
2.1 牛Ⅰ型膠原特征多肽的篩選
膠原海綿樣品經序列純胰蛋白酶酶解后產生大量有斷裂規律的低分子量多肽,將酶解后樣品用離子阱質譜(LCQ)進行分析,得到牛Ⅰ型膠原酶解多肽的總離子流圖(圖1)。利用BLAST進行膠原多序列比對,發現牛Ⅰ型膠原存在一定數量的特征多肽。利用Bioworks軟件對質譜信息用SEQUEST進行數據庫搜索,多肽篩選參數為Xcorr>1.5(單電荷),Xcorr>2.0(雙電荷),篩選出的陽性肽段見表1。然后結合手工檢查確認譜圖質量,例如離子豐度、b離子和y離子之間匹配度等。同時確保肽段短小,羥基化修飾位點低。依據以上條件,篩選出多肽GFSGLDGAK為牛Ⅰ型膠原的特征肽段。圖2和圖3分別為該肽段的提取離子流圖和質譜圖,該肽段的提取離子流圖有較高的豐度,且二級離子碎片與理論值的匹配度較高。故選擇該肽段為牛Ⅰ型膠原的特征肽段,用于牛Ⅰ型膠原的類型識別與定量。

表1 牛Ⅰ型膠原的陽性肽段信息Table 1 Positive peptide information of bovine Ⅰtype collagen
P*is hydroxyproline

圖2 特征肽段的提取離子色譜圖和一級質譜圖Fig.2 Extracted ion chromatogram and mass spectrum of marker peptide
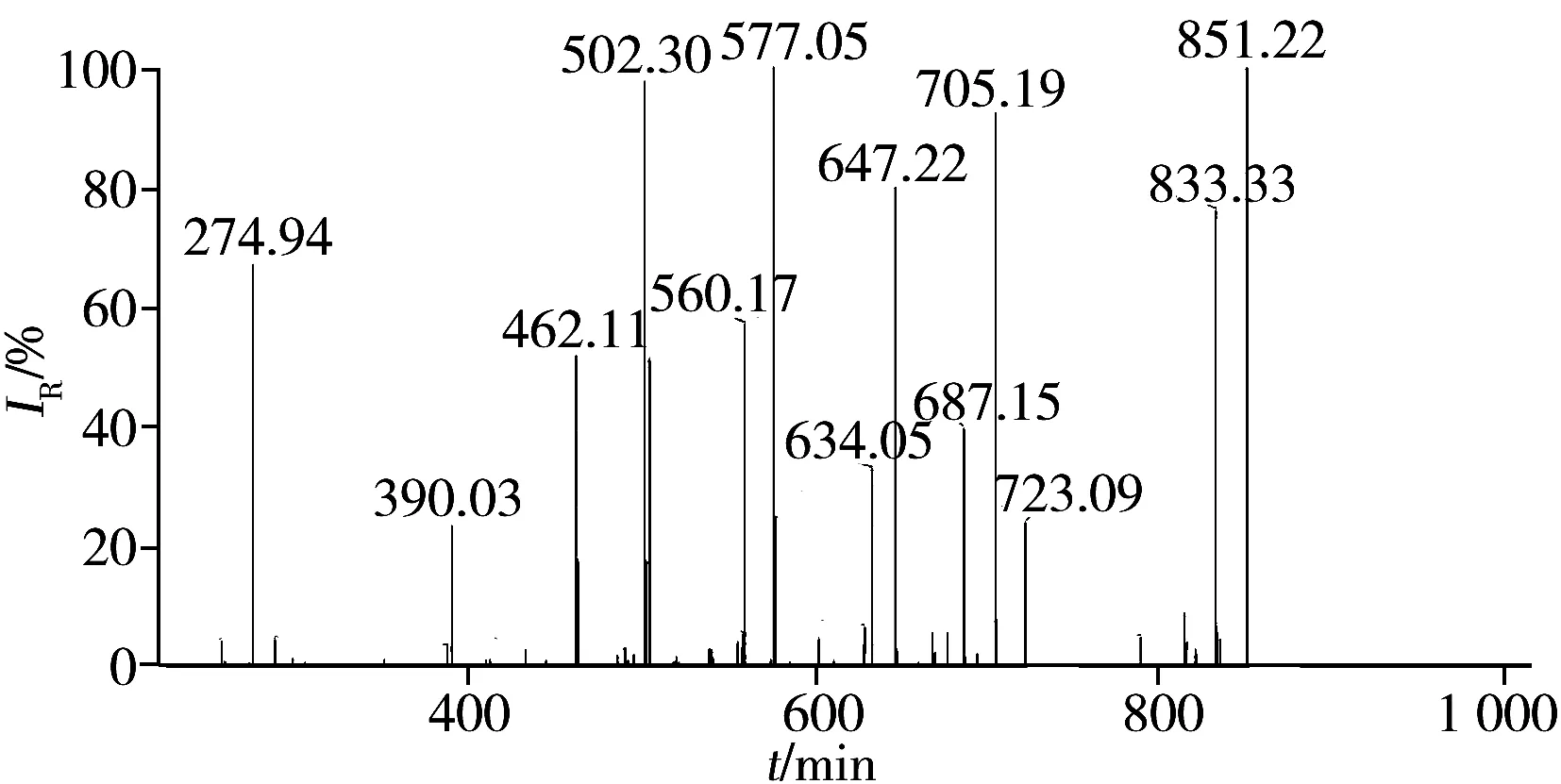
圖3 特征肽段的二級質譜圖Fig.3 MS/MS spectrum of marker peptide
2.2 樣品前處理分析
膠原海綿樣品經過2次沸水浴加熱前處理,一次是酶解前加熱使膠原解旋變性,另一次是酶解后加熱使胰酶滅活。本研究利用肽段對膠原進行定量,需確保除酶解釋放肽段外,其他步驟不會發生肽鍵斷裂。對不同相對分子質量的蛋白標準品以及膠原海綿經沸水浴加熱20 min后的樣品進行凝膠過濾分析,凝膠過濾色譜圖分別見圖4A和4B。由圖4A中不同蛋白標準品的相對分子質量與對應的保留時間計算得出,圖4B中2個色譜峰的相對分子質量分別約為276 kDa與138 kDa。文獻研究顯示,加熱后膠原變性,三螺旋解旋形成明膠,3條肽鏈完全松開或1條肽鏈松開,任意2條肽鏈由少量氫鍵連接,牛I型膠原的單條肽鏈相對分子質量為138 kDa(UniProtKB),這與本文的凝膠過濾檢測結果符合。同時圖4B中無其他雜峰,表明短時間的加熱并未使膠原肽鏈破碎,未產生低分子肽段。酶解后,利用“1.6”方法對未加熱滅活和加熱滅活的樣品進行檢測(圖5),特征肽段的峰面積相似,酶解肽未因加熱而導致進一步水解。

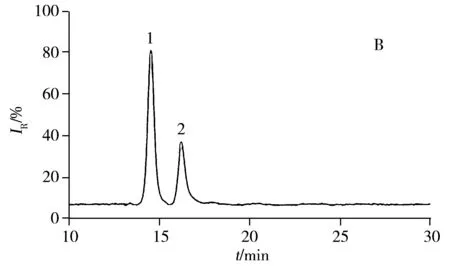
圖4 蛋白標準品(A)與膠原海綿樣品(B)的凝膠過濾色譜圖Fig.4 Gel filtration chromatograms of protein standards(A) and collagen sample(B)A.1:430 kDa,2:200 kDa,3:150 kDa,4:69 kDa,5:29 kDa;B.1:276 kDa,2:138 kDa
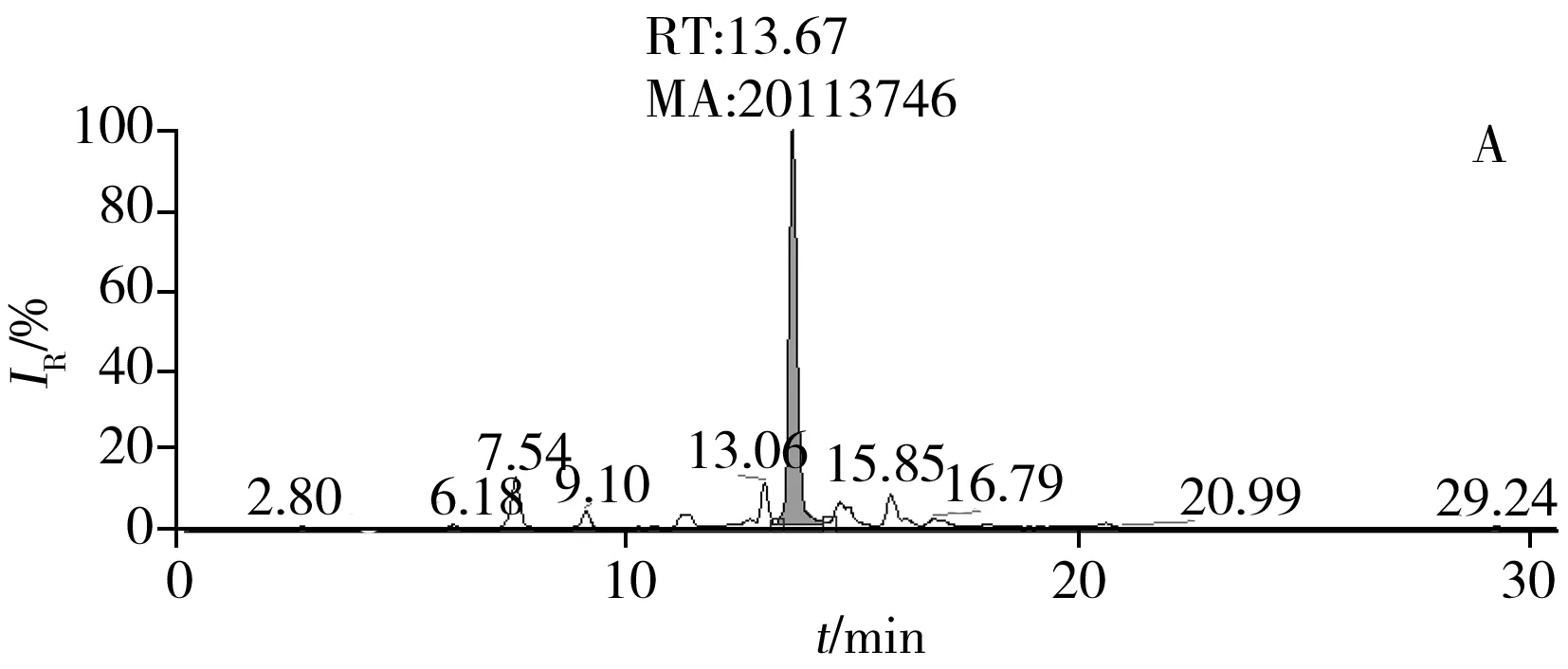

圖5 特征肽段的選擇離子監測色譜圖Fig.5 SIM chromatograms of marker peptideA:non-inactivated enzymatic sample;B:inactivated enzymatic sample
2.3 方法學驗證
2.3.1 線性范圍將特征多肽標準品稀釋至質量濃度分別為0.645、1.29、3.225、19.2、6.45、12.9、21.5 μg/mL,按照“1.6”條件進樣分析。以多肽標準品的質量濃度為橫坐標(x,μg/mL),對應的峰面積為縱坐標(y)繪制校正曲線。結果顯示,特征多肽標準品在0.645~21.5 μg/mL范圍內呈良好的線性關系,線性方程為y=6.39×109x-1.73×106,相關系數(r2)為0.999 4。
2.3.2 定量下限精密量取特征多肽標準品適量,用超純水逐步稀釋,定容,搖勻。按照“1.6”條件每個樣品進樣2針,記錄色譜圖。以信噪比為10∶1時為定量下限,測得牛Ⅰ型特征多肽標準品的定量下限為6.45×10-4μg/mL。

表2 加標回收率實驗結果Table 2 Results of the sample recovery experiment
2.3.3 回收率在1 mg/mL的膠原海綿酶解液中加入3.10、2.48、1.55 μg/mL的多肽標準品,按照“1.6”條件進樣分析,并計算回收率。由表2可知,3個加標水平下的平均回收率為98.2%~102%,相對標準偏差(RSD)在10%以內,表明本方法可行。
2.3.4 精密度取特征多肽標準品溶液,注入液相色譜-質譜聯用儀,按照“1.6”條件連續進樣6針,記錄色譜圖,計算峰面積的RSD為0.80%,說明儀器精密度良好,能夠滿足樣品含量測定需求。
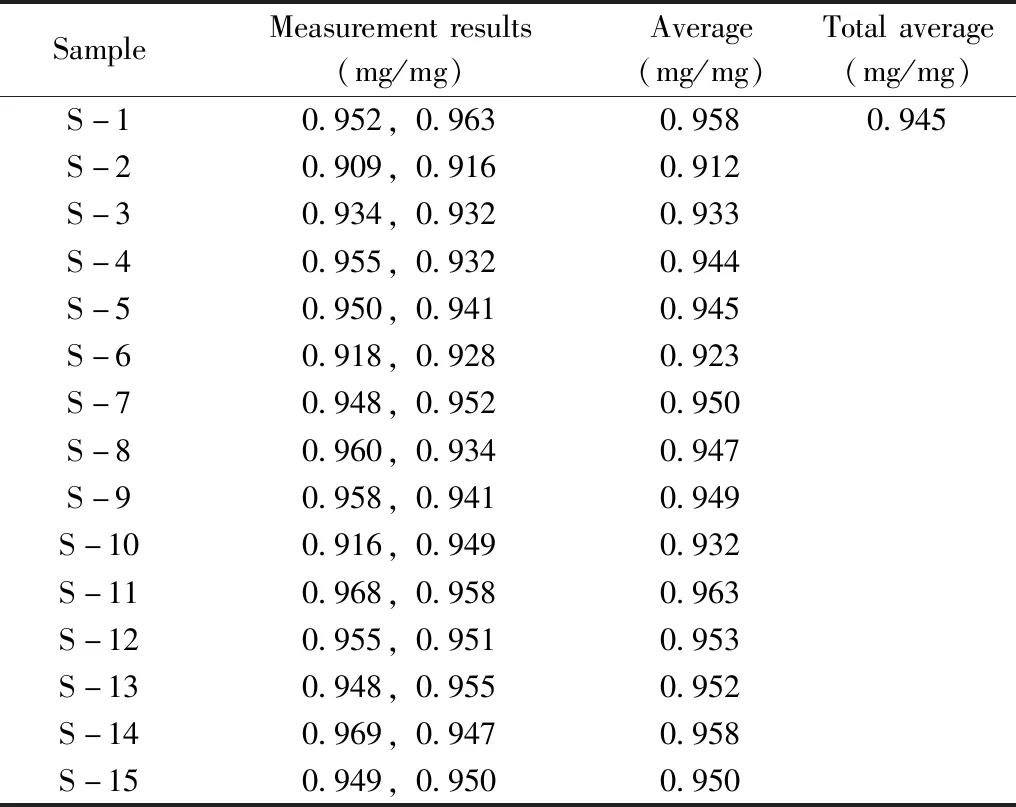
表3 膠原海綿均勻性檢驗的測量數據Table 3 Measurement data of the uniformity test of collagen standards

表4 膠原標準品的均勻性考察方差分析Table 4 Analysis of variance for collagen standard uniformity
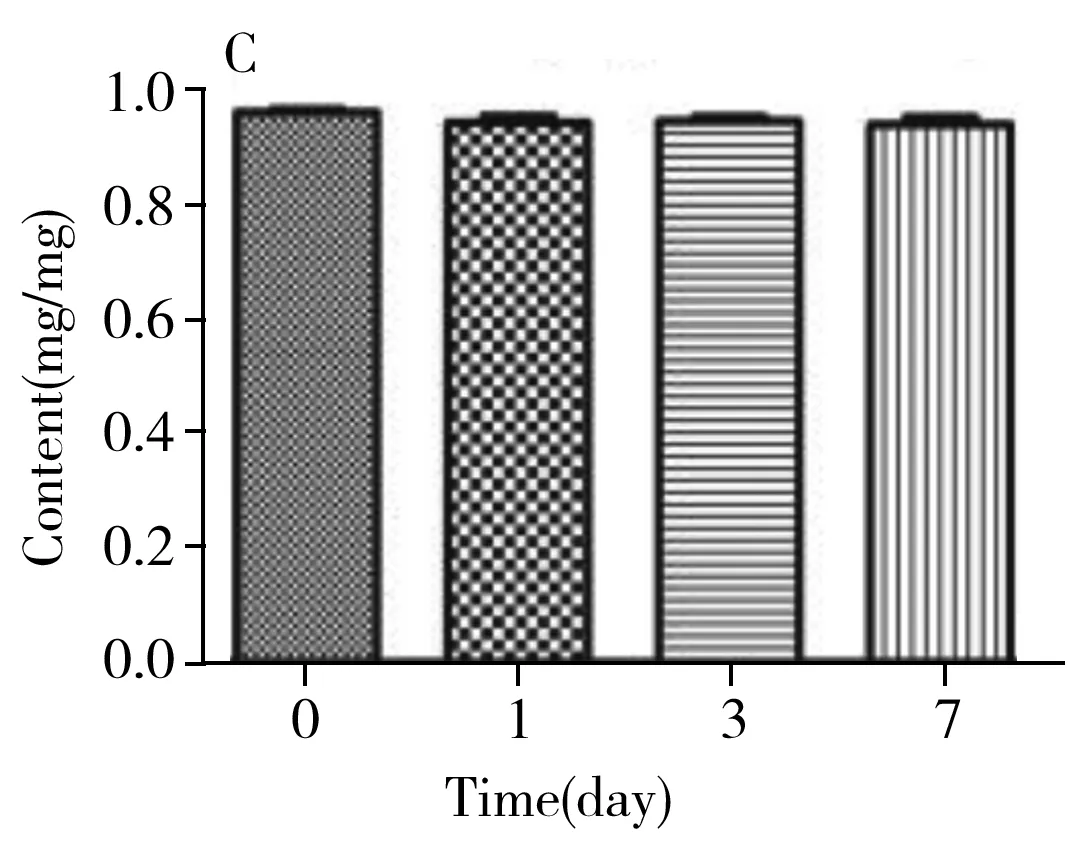
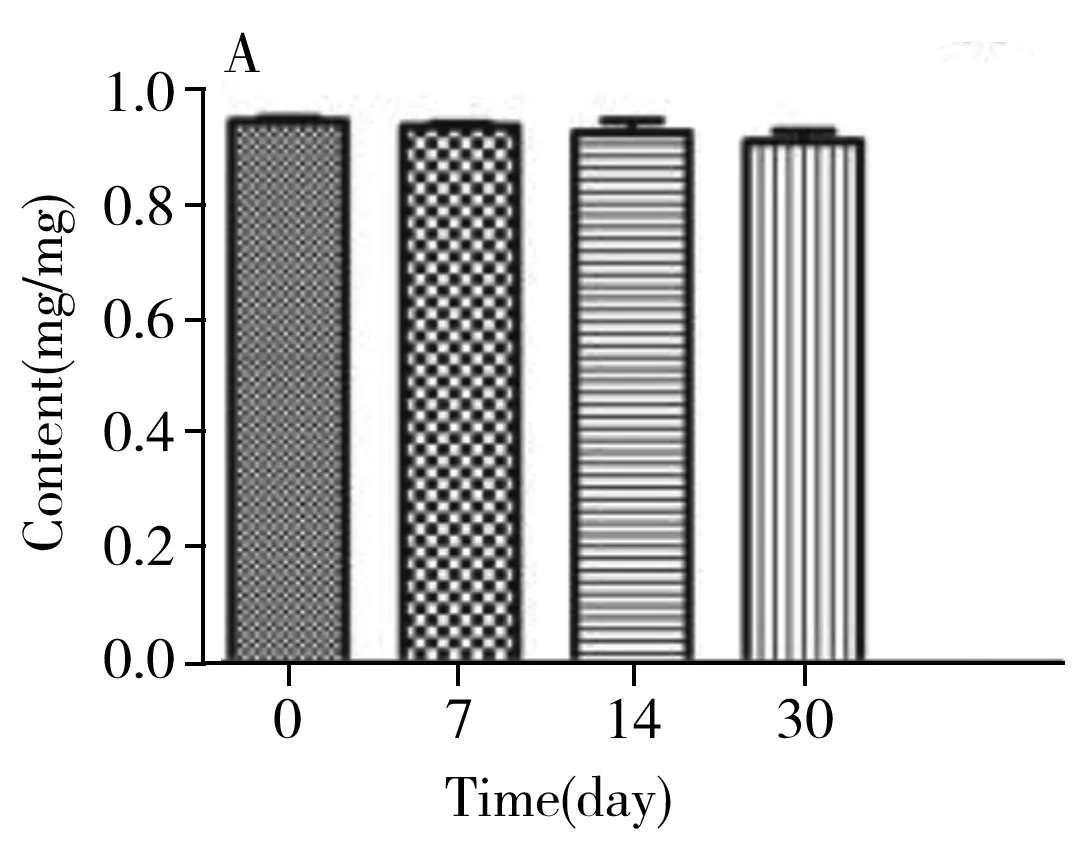
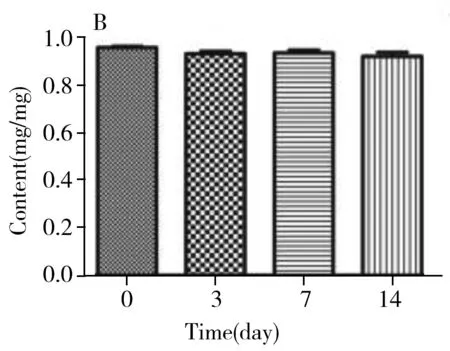
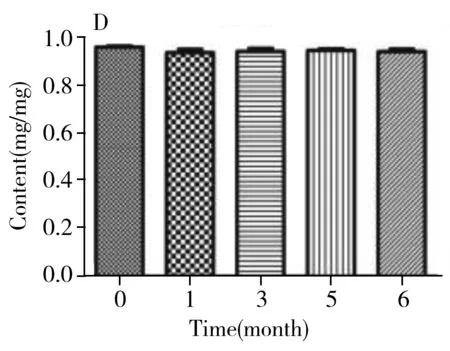
圖6 膠原海綿中牛Ⅰ型膠原含量Fig.6 Content of bovine type Ⅰ collagen in collagen spongeA:4 ℃;B:25 ℃;C:37 ℃;D:-20 ℃

表5 不同實驗室的檢測結果Table 5 Test results from different laboratories
2.4 牛Ⅰ型膠原含量檢測
2.4.1 均勻性膠原海綿樣品共210支,按順序編號后,采用EXCEL軟件生成隨機數的方法,隨機抽取15支樣品,分別編號為S1、S2……S15。每個抽取樣品按“1.6”方法重復測定2次,分析組間方差和組內方差,以評價樣品的瓶內與瓶間的均勻性。采用F檢驗,評估樣品的均勻性在單元內及單元間是否存在顯著性差異。檢測結果及方差分析結果見表3與表4。
經計算,統計量F=3.14,該統計量是自由度為(14,15)的F分布變量。根據自由度(14,15),及顯著水平α=0.01,可由F分布臨界值表查得臨界的F值為3.56。F=3.14 2.4.2 穩定性穩定性是考察在特定時間間隔和貯存條件下,物質的特性值保持在規定范圍內的能力。短期穩定性實驗的溫度分別設為4、25、37 ℃,長期穩定性實驗的溫度設為-20 ℃。每個溫度條件下貯存多支樣品,在不同時間取出3支樣品,按“1.6”方法重復測定3次。各貯存條件下的膠原含量檢測結果見圖6。對于穩定性的檢測結果通過回歸曲線方法進行判斷,穩定性評估基本模型為:Y=β0+β1X。式中:β0、β1為回歸系數;X為時間;Y為物質的特性值。 根據公式計算,4、25、37、-20 ℃下的斜率β1分別為-0.004、-0.007、-0.007和-0.004,截距β0分別為0.986、1.001、0.961和0.954,β1的標準偏差s(β1)分別為0.005、0.013、0.013和0.012。基于β1的標準偏差,用t檢驗檢測其顯著性,其中t0.95,1=12.7。在4、25、37、-20 ℃條件下,|β1| 2.4.3 定值利用本方法,聯合3家實驗室對3支膠原海綿樣品進行測定,以驗證方法的可行性與準確性,結果見表5。對該數據進行統計學分析,以判斷是否滿足定值要求并確定膠原海綿含量的標準值。首先采用t-檢驗法和狄克遜(Dixon)法對每一組獨立檢測結果進行組內可疑值檢驗,剔除可疑值,結果所有值均保留。然后采用科克倫(Cochran)法檢驗,先計算各組數據的方差,再計算其中最大方差與各組方差和之比,確保了各組數據的平均值均為等精度。隨后采用格拉布斯(Grubbs)法驗證了各組數據平均值均無顯著性差異。最后采用夏皮洛-威爾克(Shapiro-Wilk)法檢驗4組數據均符合正態分布。因此,求出的總平均值:牛Ⅰ型膠原含量=(0.926±0.014) mg/mg,為準確值。 2.4.4 水分與灰分按照“1.7”方法測定膠原海綿產品的含水量為3.24%~4.05%,灰分含量為2.58%~5.32%。 本研究建立了一種高靈敏的牛Ⅰ型膠原定性定量的方法,從特征多肽的篩選、HPLC-MS分析和數據處理等方面進行了探討。以牛Ⅰ型膠原為研究對象,篩選序列中的特征肽為外標,采用液相色譜-三重四極桿質譜進行定量檢測。在不同的加標水平下,平均回收率為98.2%~102%,峰面積的RSD為0.80%,方法具有良好的回收率和精密度。依據國家計量技術規范《標準物質定值的通用原則及統計學原理》[19]對該牛Ⅰ型膠原海綿產品的穩定性、均勻性和定值進行測定,確定該膠原海綿產品的牛Ⅰ型膠原含量為(0.926±0.014) mg/mg,證明了方法的可行性和準確性,亦證實了該產品具有成為牛Ⅰ型膠原標準品的潛力。本文為不同類型膠原的定量分析提供了可靠準確的實驗依據。3 結 論
猜你喜歡
數學小靈通·3-4年級(2024年2期)2024-05-15 02:02:28中學生數理化(高中版.高考數學)(2022年3期)2022-04-26 14:04:16數學年刊A輯(中文版)(2020年1期)2020-05-19 00:30:36空間科學學報(2020年2期)2020-04-01 03:50:40瘋狂英語·新策略(2019年10期)2019-12-13 08:43:28中等數學(2019年8期)2019-11-25 01:38:14當代陜西(2019年10期)2019-06-03 10:12:04新聞傳播(2018年11期)2018-08-29 08:15:24數學小靈通·3-4年級(2017年9期)2017-10-13 08:10:54廣西科技大學學報(2016年1期)2016-06-22 13:10:38
