HPLC-ELSD同時(shí)測定鮮竹瀝中單糖 雙糖的含量△
2020-05-19 05:34:44趙雯洪挺游媛羅躍華付輝政
中國現(xiàn)代中藥 2020年3期
關(guān)鍵詞:方法
趙雯,洪挺,游媛,羅躍華,付輝政
江西省藥品檢驗(yàn)檢測研究院 江西省藥品與醫(yī)療器械質(zhì)量工程技術(shù)研究中心,江西 南昌 330029
鮮竹瀝為禾本科植物粉綠竹Phyllostachysviridi-glaucescens(Carr.) A. et C. Riv.、凈竹PhyllostachysnudaMcClure及同屬的數(shù)種植物的鮮桿經(jīng)加熱后自然瀝出的液體,煮沸后加適量防腐劑制得,具有清熱化痰之功效,用于肺熱咳嗽痰多、氣喘胸悶、中風(fēng)舌強(qiáng)、痰涎壅盛、小兒痰熱驚風(fēng)等癥,為治療肺熱咳嗽的要藥[1-2],也是用于治療痰熱咳嗽等常見呼吸病癥中藥大品種鮮竹瀝、復(fù)方鮮竹瀝液的主要原料藥,涉及23家單方鮮竹瀝生產(chǎn)企業(yè)和10家復(fù)方鮮竹瀝液生產(chǎn)企業(yè)。鮮竹瀝主要含有愈創(chuàng)木酚、氨基酸、木脂素、無機(jī)元素、酚酸、糖等類成分[3-8]。熊艷等[9]采用柱前衍生HPLC比較了傳統(tǒng)燒制法和回流提取法2種方法制備的淡竹瀝中游離氨基酸含量,結(jié)果回流提取法所得游離氨基酸總量較燒制法高出近12倍。李紅等[10]采用紫外-分光光度法測定了竹瀝中總酚的含量,結(jié)果竹瀝中總酚質(zhì)量分?jǐn)?shù)為0.045%。熊艷等[11]采用HPLC對鮮竹瀝口服液中愈創(chuàng)木酚的含量進(jìn)行測定,該方法簡單可靠,重復(fù)性好。陳碧蓮等[12]采用高效毛細(xì)管氣相色譜法對鮮竹瀝中愈創(chuàng)木酚的含量進(jìn)行測定,該方法簡便,重復(fù)性好,靈敏度高。高吾名[13]采用原子吸收分光光度計(jì)和氨基酸分析儀對5個(gè)不同產(chǎn)地相同時(shí)令出廠的竹瀝油中6種無機(jī)元素和氨基酸進(jìn)行分析,結(jié)果竹瀝油中6種無機(jī)元素和氨基酸含量因產(chǎn)地不同差異較大,但每種無機(jī)元素占6種元素總量的比例和各種氨基酸的分布比例卻有一定的規(guī)律。筆者在對鮮竹瀝生產(chǎn)加工企業(yè)調(diào)研中發(fā)現(xiàn),不少企業(yè)使用水煮、高溫蒸等方法代替標(biāo)準(zhǔn)規(guī)定的干餾法加工鮮竹瀝,甚至有的生產(chǎn)企業(yè)直接使用蔗糖進(jìn)行勾兌,嚴(yán)重影響鮮竹瀝及制劑質(zhì)量,而鮮竹瀝收載于1992年版《衛(wèi)生部藥品標(biāo)準(zhǔn)中藥材》(第1冊),該標(biāo)準(zhǔn)項(xiàng)下有以酪氨酸為對照的薄層色譜鑒別、pH、相對密度以及總固體質(zhì)控項(xiàng)目,無指標(biāo)成分含量測定項(xiàng)[2],難以有效控制其質(zhì)量。加之鮮竹瀝制備工藝特殊,影響干餾工藝因素較多。因此,非常有必要對鮮竹瀝現(xiàn)行標(biāo)準(zhǔn)進(jìn)行提高,保障產(chǎn)品的質(zhì)量,規(guī)范市場秩序。本課題組對鮮竹瀝化學(xué)成分進(jìn)行系統(tǒng)分析,其中單糖和雙糖為鮮竹瀝中的主要特征成分,目前尚未見測定鮮竹瀝中單糖和雙糖的文獻(xiàn)報(bào)道。
本實(shí)驗(yàn)收集了按標(biāo)準(zhǔn)干餾法制備的9批鮮竹瀝和按古法直火烤的方法制備的樣品以及采用高溫蒸、水煮方法制備的非標(biāo)準(zhǔn)方法樣品,采用高效液相色譜-蒸發(fā)光散射檢測器法同時(shí)測定上述樣品中單糖和雙糖的含量,以此考察不同加工方法制備的鮮竹瀝中單糖和雙糖含量差異,經(jīng)方法學(xué)考察,該方法專屬性強(qiáng),準(zhǔn)確可行,重復(fù)性好,可作為鮮竹瀝的質(zhì)量控制方法。
1 材料
1.1 儀器
LC 20AT高效液相色譜儀(日本島津公司);Alltech3300蒸發(fā)光散射檢測器(美國奧泰公司);Sartorius BT 25S型十萬分之一電子天平(北京賽多利斯天平有限公司)。
1.2 試藥
D-果糖對照品(批號:100231-201305,純度:99.4%)、D-無水葡萄糖對照品(批號:110833-201205,純度:99.5%)、蔗糖對照品(批號:111507-201303,純度:99.8%)均購自中國食品藥品檢定研究院;乙腈為色譜純;水為娃哈哈飲用純凈水;其他試劑均為分析純。
鮮竹瀝來源:樣品1~9為銅鼓縣偉升實(shí)業(yè)有限公司生產(chǎn);樣品10~13為青春康源制藥有限公司生產(chǎn);樣品14為紅楓鮮竹瀝廠生產(chǎn);蒸(高溫)、煮、直火烤為自制樣品。
2 方法與結(jié)果
2.1 對照品溶液的制備
精密稱取D-果糖、D-無水葡萄糖、蔗糖對照品適量,加水分別制成含D-果糖162.710 46 μg·mL-1、D-無水葡萄糖211.388 4 μg·mL-1、蔗糖218.761 6 μg·mL-1的對照品溶液。
2.2 供試品溶液的制備
取鮮竹瀝樣品2 mL,置50 mL量瓶中,加水稀釋至刻度,搖勻,即得。
2.3 色譜條件
Luna?Omega SUGAR 100?色譜柱(250 mm×4.6 mm,3 μm);流動(dòng)相為乙腈-水(85∶15);流速為1.0 mL·min-1;柱溫為35 ℃,ELSD漂移管溫度為70 ℃;N2流速為2.0 L·min-1。見圖1。
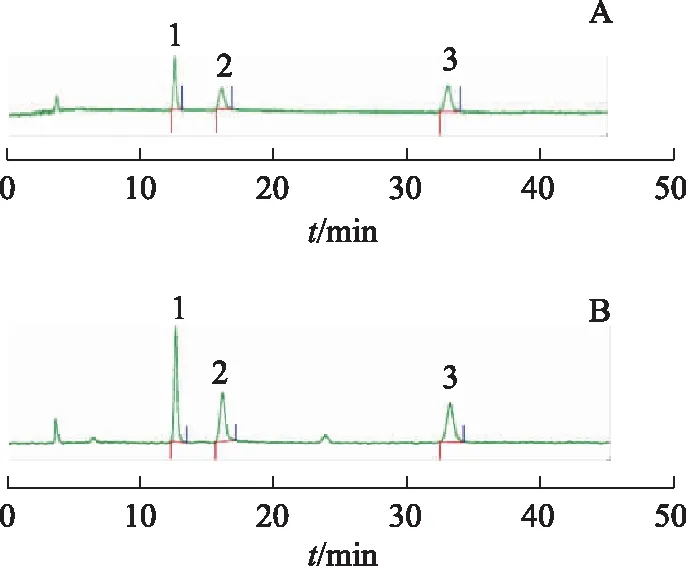
注:A.混合對照品;B.鮮竹瀝樣品;1.果糖;2.葡萄糖;3.蔗糖。圖1 混合對照品和鮮竹瀝樣品HPLC-ELSD圖
2.4 線性關(guān)系考察
取2.1項(xiàng)下的混合對照品溶液5、10、15、20、25 μL,按2.1色譜條件進(jìn)樣分析,以對照品進(jìn)樣量的對數(shù)為橫坐標(biāo),以峰面積積分值的對數(shù)為縱坐標(biāo),繪制標(biāo)準(zhǔn)曲線,結(jié)果見表1,表明各糖類成分的線性關(guān)系良好。
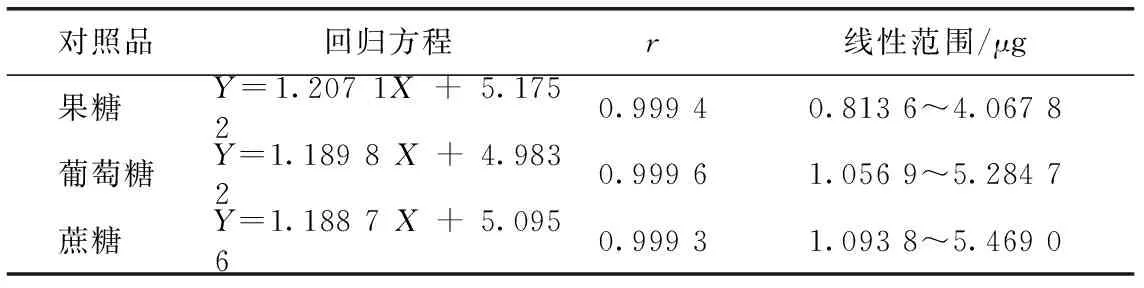
表1 線性關(guān)系考察結(jié)果
2.5 檢測限與定量限
取已知濃度的混合對照品溶液,逐級稀釋,按2.1色譜條件測定。信噪比為3∶1時(shí),測得果糖、葡萄糖、蔗糖檢測限分別為0.10、0.30、0.15 mg·mL-1;信噪比為10∶1時(shí),測得果糖、葡萄糖、蔗糖定量限分別為0.73、1.36、1.31 mg·mL-1。
2.6 精密度試驗(yàn)
精密吸取按2.2項(xiàng)下方法制備的供試品溶液10 μL,在2.1色譜條件下連續(xù)進(jìn)樣6次,測定峰面積,結(jié)果果糖、葡萄糖、蔗糖峰面積平均值分別為379 461、299 135、398 264,RSD分別為1.3%、1.7%和1.6%,表明方法精密度良好。
2.7 重復(fù)性試驗(yàn)
精密稱取同一批號(批號:20180822)的鮮竹瀝樣品6份,分別按2.2項(xiàng)下方法制備供試品溶液,在2.1色譜條件下進(jìn)行分析測定,果糖、葡萄糖、蔗糖平均質(zhì)量濃度分別為5.403 6、6.391 2、6.236 6 mg·mL-1,RSD分別為1.3%、0.9%和1.7%,表明試驗(yàn)重復(fù)性良好。
2.8 穩(wěn)定性試驗(yàn)
精密吸取同一供試品溶液(批號:20180822)10 μL,分別于0、6、12、18、24 h進(jìn)樣測定,結(jié)果果糖、葡萄糖、蔗糖峰面積平均值分別為369 903、290 412、376 556,RSD分別為1.2%、1.8%和1.6%,說明供試品溶液在24 h內(nèi)穩(wěn)定。
2.9 加樣回收率試驗(yàn)
取上述已測含量的鮮竹瀝樣品(批號:20180822) 1 mL,共6份,精密稱定,分別精密加入D-果糖、D-無水葡萄糖、蔗糖對照品適量,分別按2.2項(xiàng)下方法制備供試品溶液,精密吸取10 μL注入高效液相色譜儀,記錄色譜圖,計(jì)算平均加樣回收率,果糖、葡萄糖、蔗糖的平均回收率分別為98.1%、97.5%、99.2%,RSD分別為1.3%、0.9%、1.1%,表明方法準(zhǔn)確度符合要求。結(jié)果見表2。
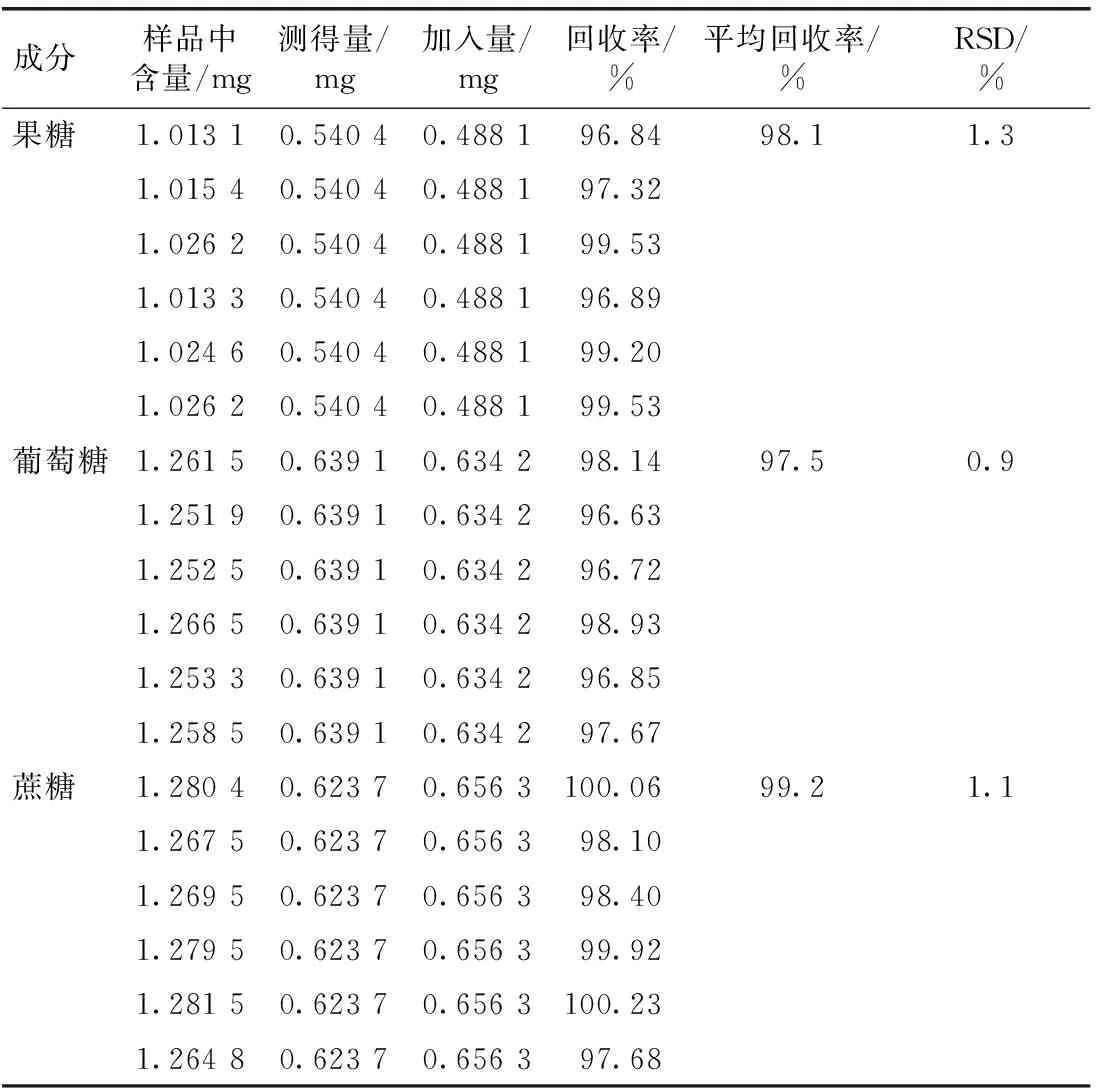
表2 鮮竹瀝3個(gè)成分的加樣回收率試驗(yàn)結(jié)果(n=6)
2.10 樣品測定
按擬訂的含量測定方法,測定17批鮮竹瀝樣品中3種糖類成分的含量,結(jié)果見表3。
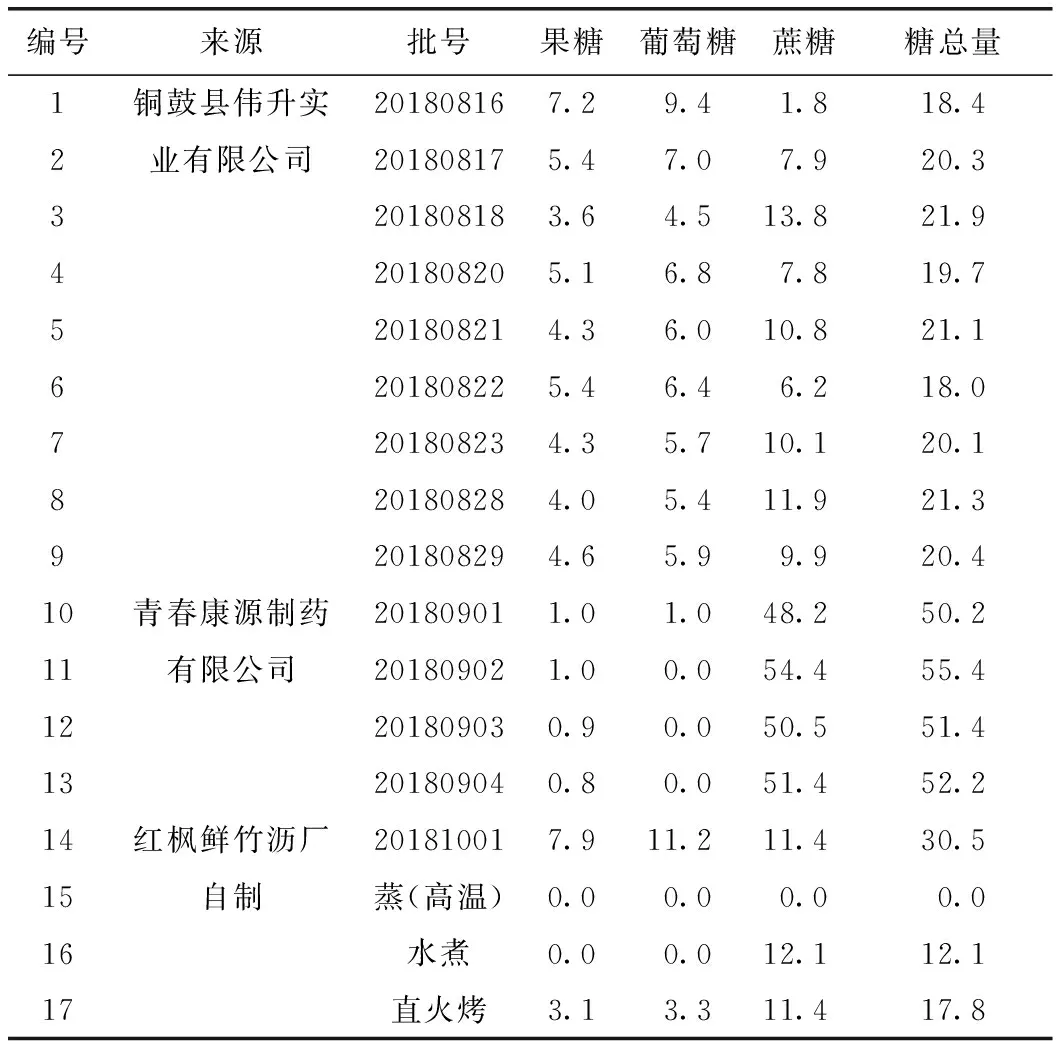
表3 樣品的含量測定結(jié)果 mg·mL-1
3 結(jié)果與討論
選用了Prevail Carbohyrate ES(250 mm×4.6 mm,5 μm)、Agilent ZORBAX NH2(250 mm×4.6 mm,5 μm)和Luna?Omega SUGAR 100?(250 mm×4.6 mm,3 μm)3根色譜柱,本次實(shí)驗(yàn)Luna?Omega SUGAR 100?(250 mm×4.6 mm,3 μm)色譜柱的分離效果最好。
比較了流動(dòng)相乙腈-水(75∶25)、乙腈-水(80∶20)、乙腈-水(85∶15)、甲醇-水(75∶25)、甲醇-水(80∶20)、甲醇-水(85∶15),結(jié)果以乙腈-水(85∶15)作為流動(dòng)相效果最好。
銅鼓縣偉升實(shí)業(yè)有限公司采用的標(biāo)準(zhǔn)干餾法與古法直火烤工藝一致。銅鼓縣偉升實(shí)業(yè)有限公司生產(chǎn)的9批樣品中糖總量為18.0~21.9 mg·mL-1,平均含量為20.1 mg·mL-1,與自制直火烤樣品中糖總量差異不大,且均檢出3種糖類成分。
青春康源制藥有限公司可能人為添加蔗糖。青春康源制藥有限公司生產(chǎn)的4批樣品中3批未檢出葡萄糖,且蔗糖含量異常高。
自制樣品中蒸(高溫)工藝制備的樣品3種糖類成分均未檢出,水煮工藝制備的樣品只檢出蔗糖,直火烤工藝制備的樣品均檢出果糖、葡萄糖、蔗糖。
4 結(jié)論
通過建立鮮竹瀝中單糖和雙糖的含量測定方法,可以對生產(chǎn)工藝蒸(高溫)、水煮以及人為添加進(jìn)行區(qū)分,具有專屬性。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(bào)(2021年2期)2021-05-25 02:07:46
中學(xué)生數(shù)理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(bào)(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
