化學腐蝕對碳化硅納米線的光致發光性能的影響
2020-05-19 08:51:48雷思潔王冬華
山東化工 2020年7期
雷思潔,王冬華
(渭南師范學院 化學與材料學院,陜西 渭南 714099)
材料對于人們的生活必不可少,隨著人們對材料要求的不斷提高以及科技的不斷發展許多新型材料也隨之出現。納米材料就是眾多新型材料中的一種。納米材料指的是在納米尺度(1~100 nm)內調控物質結構制成的具有特異性能的材料。納米材料性質上的變化是由晶體表面原子數與總原子數之比隨著粒徑變小而急劇增大而引起的。因此納米材料相比于其他材料來說有著很多優勢。碳化硅因其優異的物理、化學和電學性能、良好的強度、化學穩定性以及優異的抗氧化和抗熱震能力[1-3]。相比于常規碳化硅而言納米碳化硅受到低維數,量子限域效應以及特殊的形狀的影響,因而具有優異的力學性能,光電特性,化學惰性和生物相容性等,因此納米碳化硅被廣泛應用于半導體器件、機械制造、航天工業、生物材料及陶瓷膜等眾多領域,也是化學反應中一些催化劑載體的較理想材料。被成功的應用在一些比較重要的化學反應當中,顯示了長遠的應用前景。現如今對技術要求較低的碳化硅粗料已能大量供應,而對技術要求極高的納米級碳化硅還不能大量供給。因此對納米碳化硅的研究至關重要。
目前,已有多種制備SiC納米材料的方法,如:等離子體法[4]、溶劑熱法、模板法、氣相沉積法[5]、碳熱還原法[6]、電弧放電法[7]等。與這些方法相比,溶膠-凝膠法即能將納米材料前驅體在特定的條件下水解,生成溶膠,之后將溶劑揮發形成凝膠,再經過加熱干燥高溫熱處理等方法制備所需納米材料。張洪濤等人就采用此方法制備了高純度且含氧量低的β-SiC[8]。并且溶膠-凝膠法具有合成溫度低,反應更容易進行,產率高等特點,并能在短時間內獲得分子水平的均勻性及分子水平之上的均勻混合。且設備和實驗操作簡單,實驗成本低。因此,該方法在陶瓷、玻璃、生物材料、催化劑載體、薄膜、以及復合材料等領域得到廣泛應用。碳熱還原法是在一定溫度下用無機碳作為還原劑進行氧化還原反應的方法。這種方法可以通過改變反應物的種類、加熱方式、加熱溫度和保溫時間以及保護氣體氣流大小等參數控制反應速率,以此達到制備純度較高、粒度較小的樣品的目的。由于該方法原料成本低、實驗反應簡單,適用于大規模生產[9]。
有許多材料都能用于納米碳化硅的制備,近年來研究者們也出于各種因素的考慮,例如價格低廉、產率高、對環境污染小等,嘗試選擇用不同的碳源材料。石墨粉的主要成分為碳并且石墨粉的顆粒細小與其他做碳源的原料相比具有更易反應與反應更加完全的特點,并且石墨粉的價格低廉容易購置。以高產、高效、快捷的條件來衡量選擇石墨作為碳源十分合適。近年來研究者們已經通過不同的方法制備了不同形貌的碳化硅納米線,例如:竹節狀納米線[10]、連珠狀納米線[11]、納米彈簧[12]、項鏈狀納米線[13]等。除此之外也可將碳化硅樣品進行一定程度的刻蝕,會得到不同形貌和結構的碳化硅材料,以滿足在不同領域的特殊應用。對碳化硅的刻蝕一般有干腐蝕和濕腐蝕兩種方法。干腐蝕法即指在沒有水(包括但不限于液體水,水溶液或水蒸氣)接觸的情況下,引起的腐蝕或者氧化現象。干腐蝕法具有特殊的化學成分,因此在腐蝕過程中的選擇性低,。例如:Lauermann[14]等采用干腐蝕的方法對所制備的碳化硅進行腐蝕,并研究其反應機理;Shor[14]等也使用干腐蝕法對所制備的碳化硅進行腐蝕,使其能達到制備特殊電子裝備的要求。干腐蝕會極大的破壞原樣品的材料結構[15]。相對于干腐蝕法,濕腐蝕法(指金屬在有水存在下的腐蝕)對材料的結構破壞力小,選擇性高。因此,濕化學法被用于對常規碳化硅進行腐蝕,而獲得不同結構的納米碳化硅材料。例如:Goknur[16]等選用濕腐蝕的方法對所制備的碳化硅進場腐蝕,得到了具有規則形貌的碳化硅,并討論了腐蝕之后碳化硅納米線不同部分的腐蝕狀況。因此本實驗采用濕腐蝕的方法對所制備的碳化硅進行腐蝕。可得到結構和性能明顯不同的SiC材料。通過濕腐蝕法制備出來的納米碳化硅具有特殊形貌,有一定的潛在價值。例如:腐蝕之后的納米碳化硅能夠用于制備靈敏電感器,醫學上的探針或者捕獲一些小尺寸的分子或者納米粒子使其結構性能發生明顯改善等[17]。
本實驗采用石墨作為碳源,正硅酸乙酯(TEOS)作為硅源,硝酸鐵作為催化劑采用溶膠-凝膠法和碳熱還原法制備SiC納米材料,并利用濕化學法對所得碳化硅納米材料進行腐蝕。經過腐蝕之后發現碳化硅納米線發生了選擇性腐蝕,孿晶晶界間的立方相β-SiC碳化硅被腐蝕,但孿晶晶界卻幾乎沒有發生改變。我們對腐蝕之后的納米線進行探討,研究其中的反應機理,腐蝕前后的形貌以及納米結構的光致發光特性。
1 實驗部分
1.1 實驗藥品
無水乙醇(CH3CH2OH),成都市科龍化工試劑廠;硝酸鐵(Fe(NO3)3)、草酸(C2H2O4)、濃鹽酸(HCl),天津市天力化學試劑有限公司;氫氟酸(HF),上海化學試劑有限公司;正硅酸乙酯(TEOS),天津市科密歐化學試劑有限公司;六次甲基四胺(C6H12N4),天津市福晨化學試劑有限公司;濃硝酸(HNO3),天津市富宇精細化工有限公司;石墨粉(C),天津市登科化學試劑有限公司。
1.2 實驗儀器
X射線衍射儀(XRD日本島津6100型);紅外光譜儀(TENSORⅡ);場發射掃描電鏡(FESEM Zeiss SIGMA 500型);高溫管式爐(SGL-1600,上海大恒光學精密機械有限公司);熒光光譜儀(PLF-7000,日本日立公司);箱式電阻爐(上海錦凱SXJK8-13);恒溫干燥箱;集熱式恒溫加熱磁力攪拌器;電子天平。
1.3 碳化硅的制備
第一步:用稱量紙在稱量天平上稱取15 g石墨備用,分別配置濃度為3.4%的草酸溶液和濃度為38.5%的六次甲基四胺溶液備用。
第二步:用稱量紙稱取3 g硝酸鐵放入燒杯中加入100 mL無水乙醇后用磁力攪拌器攪拌直到硝酸鐵完全溶解再加入50 mL正硅酸乙酯,用磁力攪拌器攪拌均勻后逐滴加入10 mL草酸溶液,用保鮮膜密封再均勻攪拌30 min(轉速為600 r/min)。
第三步:向上述溶液中加入15 g石墨粉,再加入100 mL無水乙醇,用磁力攪拌器密封攪拌24 h后,逐滴加入10 mL六次甲基四胺,密封攪拌至凝膠狀態,把保鮮膜打開一段時間,將得到的凝膠放入干燥箱中干燥7~8 h(溫度為110℃烘干為止)。
第四步:將第三步得到的干凝膠樣品研磨成粉裝入瓷舟中放入通有氬氣為保護氣的管式爐中煅燒12 h(設定溫度由室溫升至反應溫度1350℃用時5 h;1350℃恒溫5 h;最后溫度從1350℃降到500℃用時2 h等到溫度降到500℃后關閉管式爐。在此過程中當溫度上升到1000℃時要適當加大保護氣流)。
第五步:待樣品在爐內冷卻至室溫后取出,移至馬弗爐中繼續進行煅燒,將爐溫設置為800℃(升溫速率設置為20℃/min)持續煅燒2 h然后在爐內冷卻至室溫后取出,接著將樣品置于塑料瓶中,用濃鹽酸和氫氟酸為1∶3的比例酸化24 h,將酸化過后的樣品用大量去離子水抽濾至中性,放入干燥箱中烘干。
第六步:重復第五步的操作步驟,等到樣品完全冷卻后,將樣品裝入干凈的試劑瓶中,最終得到淺綠色的SiC樣品。
第七步:將所得的一部分SiC樣品放入塑料瓶中,用濃硝酸和氫氟酸比例為1∶3的比例腐蝕(在此過程中先加入濃硝酸)并將塑料瓶置于水溫為80℃的水浴鍋中腐蝕30 min。將腐蝕過后的樣品用大量去離子水抽濾至中性,放入干燥箱中烘干。等到樣品完全冷卻后,將樣品裝入干凈的試劑瓶中,最終得到顏色更淺的SiC樣品。
1.4 產物的表征
1.4.1 X射線衍射分析(XRD)
通過XRD對制備的樣品進行晶體衍射從而對其進行物質分析,以測定所制備的碳化硅樣品純度。測定條件為:銅靶,掃描速度為5°/min,2θ角掃描范圍為10~80°,步長為0.02°。
1.4.2 場發射掃描電子顯微鏡(FESEM)
用掃描電子顯微鏡對腐蝕前后的碳化硅樣品進行測定,因為掃描電鏡的放大倍數約為原信號的20萬倍左右,能對樣品進行形貌,微區分析和晶體結構等多種微觀組織結構信息的同位分析。通過掃描電鏡的分析我們能清楚的觀察到腐蝕部分的分布情況與腐蝕之后的形貌。
1.4.3 紅外光譜分析
因為不同化學鍵對紅外的吸收程度不同,用紅外測得光譜后與標準圖譜進行對比可研究碳化硅樣品腐蝕前后化學鍵是否出現了變化。
2 結果與討論
2.1 X射線衍射分析
SiC樣品腐蝕前后均為淺綠色粉末,但腐蝕之后的顏色較淺。由圖1可以得出,衍射譜圖中各峰對應的2θ值分別為35.6°、41.4°、60.0°、71.8°、75.6°。這五個較強的衍射峰與立方晶型β-SiC的(111)、(200)、(220)、(311)、(222)的衍射面所對應[18],并且SiC樣品腐蝕前后所測得的XRD圖譜具有相同的特征峰并且沒有其他特征峰出現,說明所制備的SiC樣品腐蝕前后均為純凈物并無其他雜質存在。
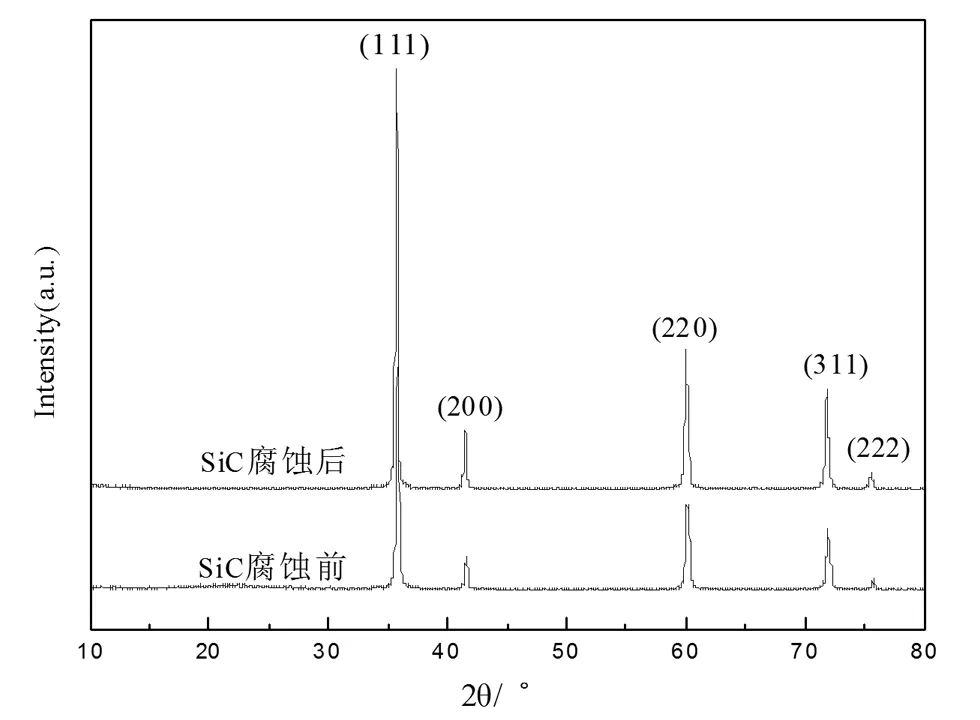
圖1 腐蝕前后SiC樣品的XRD譜圖
2.2 樣品形貌分析
如圖2所示圖中(a)、(b)表示的是未腐蝕之前的SiC樣品的場發射掃描電鏡照片。從圖中可清晰的觀察到納米SiC樣品為不規則團聚在一起的蠕蟲狀的納米線。納米線的直徑在60~100 nm之間,長度為幾個微米。而且每條納米線的方向存在差異;還可以看出納米線表面有凸起的現象。圖(c)、(d)表示的是腐蝕之后的SiC樣品的場發射掃描電鏡圖。從圖中可以清楚的觀察到腐蝕之后的納米SiC樣品仍為蠕蟲狀的納米線且存在團聚現象,長度處于1~8 μm之間。在電鏡下可清楚的觀察到納米線被酸腐蝕后出現了明顯的片狀結構,片層的直徑約為幾十納米,片層厚度約為3 nm,片層之間的部分直徑明顯減小,這說明樣品發生了選擇性腐蝕,其原因可能為:
SiC + 2HNO3+2H2O → SiC4++ 4OH-+ 2HNO2(1)
SiC4++ 4OH-→SiO2+ CO2+ 2H2(2)
SiO2+ 6HF → H2SiF6+ 2 H2O (3)
總反應式為:SiC + 2HNO3+ 6HF → H2SiF6+ 2HNO2+ CO2+ 2H2(4)
在80℃的硝酸環境中,完好晶型的β-SiC表面被氧化,生成二氧化硅,同時放出二氧化碳及氫氣,而二氧化硅則被氫氟酸溶解[16]。而晶體缺陷部分由于具有較強的抗腐蝕能力,在混合酸中并未被腐蝕,因此經過混合酸的腐蝕后出現了圖中的結構。

圖2 腐蝕前后SiC樣品的掃描電鏡圖
2.3 紅外光譜分析
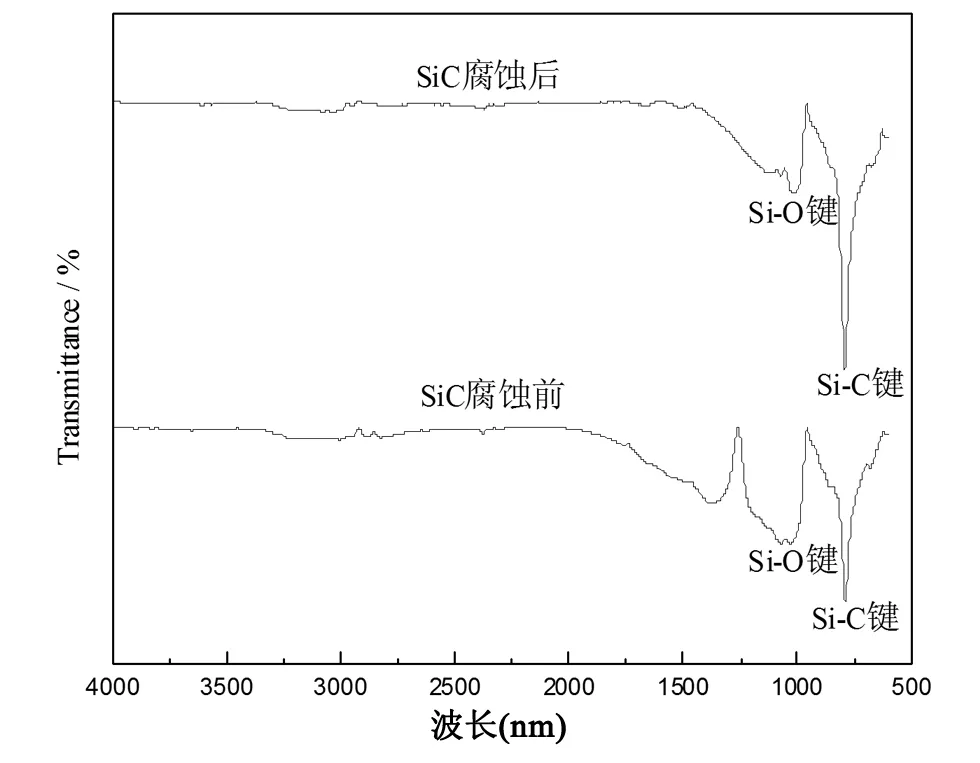
圖3 腐蝕前后SiC樣品的紅外光譜圖
圖3是SiC納米材料在混合酸腐蝕前后的紅外譜圖,從圖中可以清楚的發現在770~970 cm-1附近腐蝕前后皆存在明顯的特征吸收峰,此特征峰是由于Si-C鍵的振動吸收而產生的。因為樣品經酸腐蝕后,一部分Si-C鍵被破壞,所以腐蝕后的特征峰值較小。在1000~1100 cm-1也存在一個特征峰,此峰為Si-O鍵的特征吸收峰,腐蝕前Si-O鍵的出現是由于納米SiC顆粒直徑較小,具有較高的表面能和比表面積,且在煅燒過程中極易被氧化形成SiO2薄膜[19]。腐蝕后Si-O的出現是由于反應過程中有SiO2生成,很大一部分與HF酸發生反應,但反應不完全,所以Si-O吸收峰強度較小。由圖觀察可得到在1100 cm-1處有一個弱的吸收峰,此峰是由于Si-O鍵的不對稱伸縮振動引起的[20]。在3500 cm-1附近出現因物理吸附水而產生的-OH鍵特征吸收峰。
2.4 光致發光性能
圖4 中的(a)、(b)為不同激發波長下SiC納米材料腐蝕前后的光致發光圖譜。從圖中可得出,在相同激發波長下,樣品腐蝕前后的發射中心發生了變化。對比在相同激發波長下SiC納米材料腐蝕前后的光致發光圖譜,可以得出,兩者都有十分明顯的發射峰出現,且發射峰的位置及發射峰強度有明顯差異。由于腐蝕前的SiC樣品具有良好的晶型,所以腐蝕前的SiC發射峰的強度較大。一些材料科學家認為:納米碳化硅晶體的形貌不同,光致發光特性會有較大的區別[21-22]通過圖譜對比可知,影響SiC光致發光特性的主要因素是樣品的結構缺陷。原因是混合酸腐蝕掉了碳化硅納米中的完整晶體,導致碳化硅納米線由完整結構變為片狀結構。從表1可以看出當激發波長由290 nm增加到340 nm時,腐蝕前發射峰的中心位置由375 nm減小到367 nm,腐蝕后發射峰的中心位置也由381 nm減小到370 nm。由數據分析可知,SiC樣品腐蝕前后均發生了藍移現象,但腐蝕后的藍移現象更為明顯。這可能是由所制備的樣品缺陷引起的[23]。從表2可以看出當激發波長由290 nm增加到340 nm時,腐蝕前發射峰的強度由0.357增加到0.943。腐蝕后發射峰的強度由0.299增加到0.924。由數據分析可知,SiC樣品腐蝕前后的發射峰強度都是隨著激發波長的增加而增加的。腐蝕前的發射峰強度均大于腐蝕后的發射峰強度。這是由于腐蝕后一部分SiC被破壞,導致發射峰的強度降低。
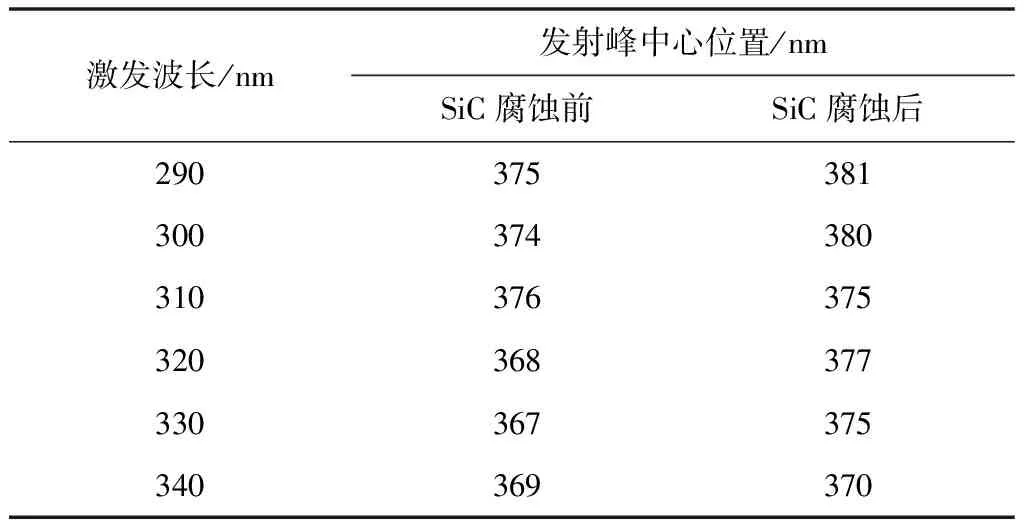
表1 腐蝕前后SiC樣品不同激發波長下發射峰的中心位置
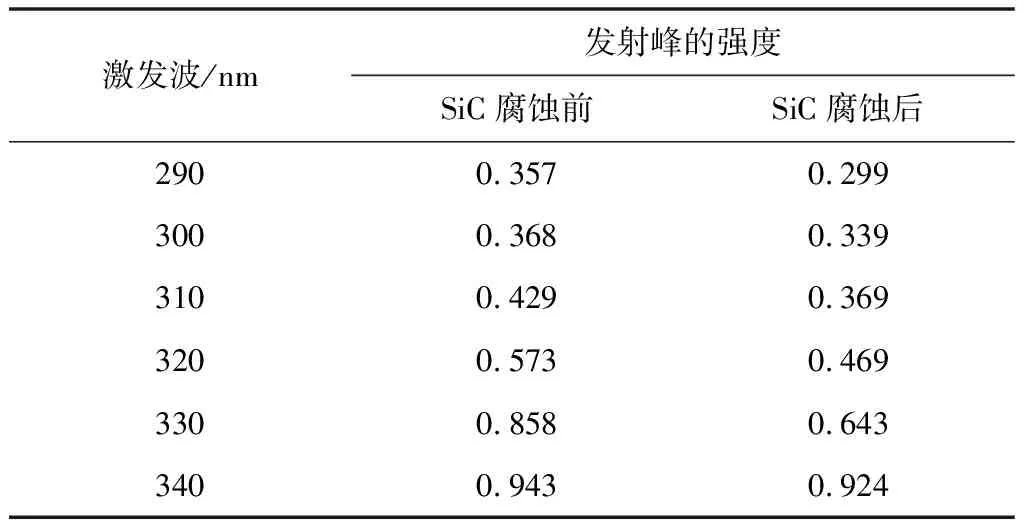
表2 腐蝕前后SiC樣品不同激發波長下發射峰的強度
3 結論
(1)本實驗以石墨粉作為碳源,正硅酸乙酯作為硅源,采用溶膠-凝膠法與碳熱還原法來制備SiC納米材料。用比例為1∶3的濃硝酸和氫氟酸對所制備的樣品進行腐蝕。
(2)使用XRD檢測得到SiC納米材料發現其特征峰明顯,且無其他雜峰出現,表明制備的樣品為純凈的SiC納米材料。對樣品形貌進行分析發現酸腐蝕前后SiC的形貌差異大,納米線的直徑與長度有明顯變化。
(3)通過對發射峰中心位置的分析,得出造成SiC光致發光性能的主要原因是晶體的結構缺陷。結構缺陷較多時,會使納米材料表面滿足量子尺寸效應發生的條件,從而使發射峰的中心位置發生明顯的藍移。
