雪菊中木犀草苷的富集及近紅外光譜分析方法
2020-05-19 15:23:37楚剛輝王坤尹學博
分析化學 2020年4期
楚剛輝 王坤 尹學博
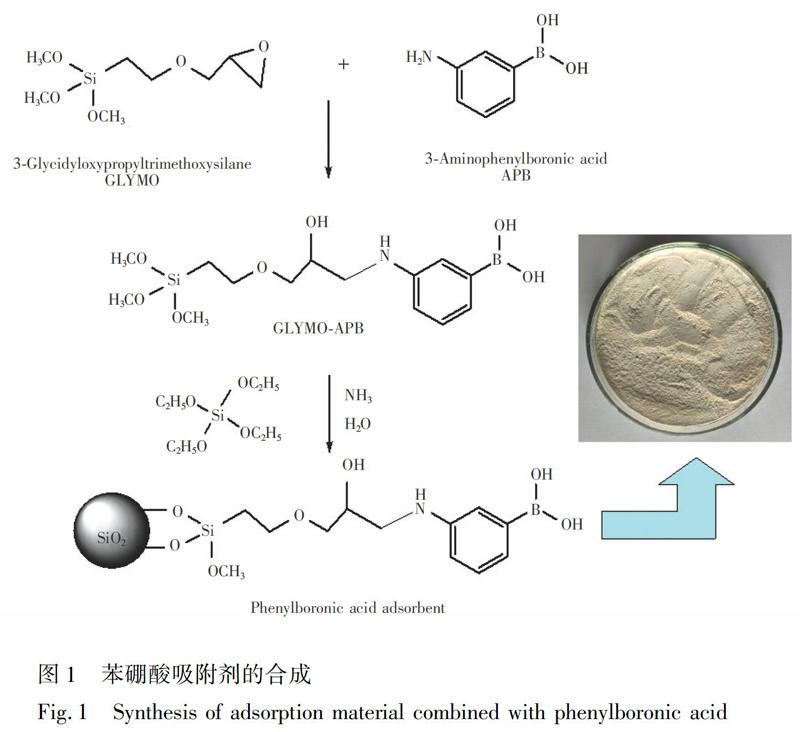


摘?要?采用溫和的方法制備了苯硼酸吸附劑,將其用于雪菊中微量木犀草苷的富集,并以此為基礎建立了快速、選擇性地測定雪菊中木犀草苷含量的近紅外光譜分析方法。富集了木犀草苷的吸附劑無需脫附,即可用近紅外漫反射光譜直接檢測,通過偏最小二乘回歸法建立了定量分析木犀草苷的校正模型,并成功實現了木犀草苷含量的測定。采用此苯硼酸吸附劑0.2 g,常溫振蕩吸附20 min后,吸附效率可達85.5%。結果表明,經連續小波變換處理近紅外光譜分析數據,木犀草苷預測模型的預測濃度和參考濃度之間的相關系數為0.9765,且木犀草苷在0.15~19.5 mg/L濃度范圍內具有較好的預測結果,預測回收率為84.7%~113.2%。本研究建立了苯硼酸吸附劑吸附預富集方法,與近紅外漫反射光譜結合,實現了雪菊溶液中微量木犀草苷的選擇性測定,為近紅外光譜與吸附富集分析藥食用植物中的其它有效成分的研究提供了參考。
關鍵詞?木犀草苷;苯硼酸吸附劑;富集;選擇性檢測;近紅外漫反射光譜
1?引 言
木犀草苷(Luteoloside)是一種天然植物黃酮類物質,為弱酸性四羥基黃酮類化合物。木犀草苷又名木犀草素-7-O-葡萄糖苷(Luteolin-7-O-glucoside),其苷元為木犀草素,因最初是從木犀草科木犀草屬植物木犀草中分離出而得名。木犀草苷具有抗炎、抗病毒、抗腫瘤和降低膽固醇等藥理作用[1,2]。雪菊是一種雙色堅果菊花,作為一種藥食兩用植物,藥理研究證實雪菊具有極高的藥用價值。雪菊的有效成分包括黃酮類、糖類、礦物質元素、酚類等,具有清熱解毒、活血、養胃、健脾、抗疲勞和抗氧化等功效[3~5]。植物樣品和中藥中木犀草苷的常見檢測方法包括高效液相色譜法[6,7]、超高效液相色譜-質譜聯用法[8]、高效液相色譜-質譜聯用法[9]和超臨界色譜法[10]等。這些方法選擇性好,但不同程度上存在有機試劑消耗量大、操作過程繁瑣、分析時間長等問題。
近紅外光是介于中紅外和可見光之間的電磁波,其波長范圍為780~2526 nm[11],近紅外光譜技術可根據有機物含氫基團(主要包括CH、NH、OH、SH等)對近紅外光譜吸收的差異,區分不同物質并測定其濃度[12],具有無損、簡單、方便、處理量大、快速、準確、環境友好等優點[13]。自20世紀90年代以來,近紅外光譜技術逐漸應用在生產和基礎研究中 [14,15]。藥食兩用植物屬于復雜體系,選擇性檢測其中的有效成分一直是相關研究的熱點,而近紅外光譜可用于復雜體系成分分析[16,17]。由于近紅外光譜的檢出限高,待測組分的含量一般應大于0.1%。為提高近紅外光譜的檢出限和分析靈敏度,文獻報道了多種吸附材料,用于樣品中微量成分的富集及近紅外光譜檢測[18~20]。為了提高檢測的靈敏度和選擇性,本研究基于含苯硼酸基團的化合物與多元醇化合物的共價化學鍵特異性結合的性質[21,22],采用溫和的方法制備了苯硼酸吸附劑,用于富集雪菊中微量的木犀草苷,采用近紅外光譜結合化學計量學方法實現了其中木犀草苷的選擇性檢測。
2?實驗部分
2.1?儀器與試劑
SF-TDL-2SOA低速離心機(上海菲恰爾分析儀器有限公司);SJZ-82電熱恒溫水浴鍋(天津市泰斯特儀器有限公司);DZF-6050真空干燥箱(上海齊欣科學儀器有限公司);HY-2型多用調速振蕩器(江蘇省金壇醫療儀器廠);AntarisⅡ近紅外光譜儀(美國Thermo公司);TU-1900 型雙光束紫外可見分光光度計(北京普析通用儀器有限公司);DF-4型壓片機(天津市港東科技發展有限公司);JJ-2增力電動攪拌器(金壇市醫療儀器廠)。
3-氨基苯硼酸(3-Aminophenylboronic acid,APB)、3-縮水甘油基氧基丙基三甲氧基硅烷(3-Glycidyloxypropyltrimethoxysilane,GLMYO)、硅酸四乙酯(Tetraethyl orthosilicate,TEOS)、木犀草苷(上海阿拉丁生化科技股份有限公司);甲醇、氨水和無水乙醇均為分析純試劑。選擇產于新疆巴楚的雪菊,經粉碎機粉碎后備用。
2.2?實驗方法
2.2.1?苯硼酸吸附劑的制備?參考文獻[23]的方法,并做適當修改。取1.16 g APB、80 mL甲醇及2.0 mL GLYMO于三頸圓底燒瓶中,在40℃恒溫水浴中電動攪拌反應12 h,得到GLYMO-APB。然后加入6.0 mL TEOS、2.0 mL純凈水、2.0 mL氨水,繼續電動攪拌24 h(圖1)。GLYMO-APB和TEOS在氨水條件下同步水解完成合成過程。反應完畢,將產物離心,先用水洗至中性,再用甲醇洗滌一次,收集固體樣品,于真空干燥箱中60℃烘干24 h,干燥后研細,備用。
2.2.2?木犀草苷的吸附實驗?以木犀草苷為研究對象,稱取0.20 g苯硼酸吸附劑于100 mL錐形瓶中,加入50.0 mL木犀草苷/雪菊提取溶液,在室溫下,于振蕩器中振蕩20 min,經量筒式過濾器過濾,測定濾液在349 nm的吸光度,木犀草苷的吸附率(R,%)的計算公式如下:
式中,A0和A分別代表木犀草苷溶液吸附前后的吸光度。
2.2.3?木犀草苷的HPLC測定法?采用HPLC法進行供試雪菊樣品中木犀草苷的含量測定。 色譜條件: Agilent HC-C18色譜柱(250 mm ×4.6 mm,5 μm),流動相A為乙腈,B為0.2%醋酸溶液,梯度洗脫: 0~40 min,15%~90% A;40~50 min,90%A。流速: 1 mL/min;柱溫,常溫;進樣量: 10 μL;檢測波長: 349 nm。配制0.5~590 mg/L木犀草苷系列標準溶液,分別取10 μL,按上述色譜條件分析,獲得木犀草苷色譜峰的峰面積。以峰面積為縱坐標,質量濃度(mg/L)為橫坐標,得線性回歸方程: y=76330x-2×106(R2=0.9820)。 雪菊樣品溶液采用微孔濾膜(0.45 μm)過濾,取續濾液,進行色譜分析,木犀草苷標準樣和雪菊提取液的色譜圖見圖2。
2.2.4?雪菊分析試液的制備與木犀草苷的富集?分別稱取適量的巴楚雪菊于100 mL錐形瓶中,加入50 mL 35% (V/V)甲醇浸泡過夜(12 h);抽濾,收集上清液于干凈錐形瓶中,備用。按照雪菊樣品色譜檢測的濃度結果,制成系列不同木犀草苷濃度的雪菊提取液68組,濃度范圍為1.5~19.5 mg/L。稱取0.2 g苯硼酸吸附劑于100 mL一系列錐形瓶內,將制備的68組雪菊提取溶液每組50 mL依次加入其中。于常溫下振蕩20 min,實現木犀草苷的富集,抽濾得68個富集有木犀草苷的吸附劑固體。富集有木犀草苷的吸附劑無需脫附,直接用于近紅外光譜檢測,得到68個樣本的近紅外光譜,其中39個用于建立定量校正模型,29個(其中12對重復樣本)作為預測模型建立近紅外光譜分析方法。
2.2.5?光譜測量方法?68個固體樣本通過近紅外積分球漫反射測量得到近紅外光譜,選擇儀器分辨率為4 cm1,光譜掃描范圍為4000~10000 cm1。于室溫下開機預熱1 h后進行測量,測量時,每隔1 h,背景校正1次。為了減小誤差,68個樣本進行隨機測量;為提高樣品測定的信噪比,每個樣品近紅外光譜均經過64次掃描,并重復3次測量,以3次結果的平均值作為最終光譜。本研究聯合多變量校準法和偏最小二乘回歸(Partial least square regression,PLSR)進行分析,并根據儀器軟件包TQ analyst 9實現建模和預測。
3?結果與討論
3.1?苯硼酸吸附劑的紅外光譜
為驗證苯硼酸的成功接枝,將二氧化硅和苯硼酸吸附劑與KBr按質量比1∶100~1∶200壓片,紅外光譜見圖3。通過比較二氧化硅的紅外光譜,苯硼酸吸附劑在1357 cm?1顯示BO鍵的伸縮振動峰,1506和1598 cm?1處有苯環的特征吸收峰,2943和2880 cm?1處屬于甲基和亞甲基的伸縮振動峰。與二氧化硅紅外光譜比較,這些新出現的吸收峰表明苯硼酸已經結合在二氧化硅表面。
3.2?苯硼酸吸附劑對木犀草苷的吸附
為確定苯硼酸吸附劑對木犀草苷的吸附性能,以10 mg/L 木犀草苷標準溶液為目標溶液,按照2.2.2節的方法進行吸附實驗。采用紫外-可見光譜檢測吸附前后木犀草苷溶液的光譜,計算吸附率(圖4),其中349 nm為木犀草苷的特征吸收波長。研究表明,苯硼酸可與含有順式二醇結構的鄰苯二酚類物質發生特異性結合作用[24,25]。比較10 mg/L木犀草苷標準溶液的吸收光譜(a曲線)和經苯硼酸吸附劑吸附后的吸收光譜(b曲線),349 nm處的吸收明顯減少,計算得到木犀草苷吸附率為85.5%。結果表明,苯硼酸吸附劑對木犀草苷具有良好的吸附作用。
按照2.2.4和2.2.5節的方法對吸附了木犀草苷的吸附劑進行光譜檢測,得到了68個固體樣品的近紅外光譜(圖5)。根據儀器軟件包TQ analyst 9,選擇的波數范圍為4115~8115 cm?1。
3.3?木犀草苷的近紅外光譜定量結果
3.3.1?木犀草苷定量校正模型的建立?通過PLSR法建立木犀草苷定量模型,為了優化定量校正模型,選擇不同的方法,如一階導數(First derivative,1st)、標準正態變換(Standard normal variate,SNV)、多元散射校正(Multiplicative scatter correction,MSC)、連續小波變換(Continuous wavelet transform,CWT)、移動窗口最小二乘多項式平滑(Savitzky-golay smoothing,SG)等光譜預處理方法[26,27],通過蒙特卡洛交叉驗證(Monte carlo cross-validation,MCCV)法確定模型的因子數,表1為不同光譜預處理方法得到的木犀草苷PLSR定量校正模型及交叉驗證結果。這些模型的因子數在8~12之間,因為近紅外光譜中既包括了木犀草苷的信息,也包含有苯硼酸吸附劑和雪菊中其它復雜化合物的組分信號。采用PLSR方法,在樣品中提取木犀草苷的定量信息,以3個參數綜合評價木犀草苷PLSR定量模型的預測能力,分別為相關系數(Related coefficient,R)、交叉驗證均方根誤差(Root mean square error of cross validation,RMSECV)和預測殘差值(Residual predictive deviation,RPD)。R反映木犀草苷的預測濃度與真實濃度的相關程度,RMSECV反映預測濃度與實際濃度的偏離程度。前期文獻結果表明,當模型的RPD>2.5時,該處理方法可用于定量預測,RPD越大,說明定量結果越準確[28,29]。
由表1可知,相比無處理方法,雪菊樣品的近紅外光譜經二階導數(Second derivative,2nd)、MSC、SNV處理后,RPD值顯示結果更差;而經1st、SG平滑、CWT、MSC+1st、SNV+1st處理后,RPD值更大,說明這些光譜預處理可有效改善定量結果。比較這5種方法與無處理方法中的R、RMSECV、RPD值,經CWT處理后的相關系數R值最大,且RMSECV值最小,RPD值最大,達到5.88。為了使光譜干擾更小、誤差更小、預測更準確,達到最優的定量效果,本研究選用CWT方法對所有木犀草苷樣品的近紅外光譜進行處理。結果表明,通過預富集和CWT方法可以提取樣品中待測組分木犀草苷的有效檢測信息。
3.3.2?木犀草苷定量模型的驗證?使用29個預測集樣品(12對重復樣本)檢驗所建立的PLS模型,以考察對木犀草苷定量校正模型的預測能力。預測集樣品的制備方法、光譜測量條件、建模方法類似于校正集樣品。圖6中的虛心圈為校正集樣品的散點,實心圈為預測集樣品的散點,虛線為校正集樣本的擬合曲線,實線為預測集樣本的擬合曲線。由圖6可見,模型的擬合值均勻分布在被擬合值點附近,兩類樣品偏PLS模型中預測濃度與參考濃度之間的擬合關系良好。在0.15~19.5 mg/L濃度范圍內,測得預測集樣品的相關系數為0.9765,預測均方根誤差(Root mean square error of prediction,RMSEP)為0.9745 mg/L。木犀草苷預測集樣本的回收率在84.7%~113.2%范圍內,表明能夠實現雪菊提取液中木犀草苷的富集,為近紅外光譜的微量檢測提供了可能。綜上,雪菊提取液中木犀草苷經苯硼酸吸附劑后,即使存在有各種基體干擾,仍能被準確測定,因此可實現雪菊中木犀草苷的選擇性檢測。模型診斷一般用殘差的結果進行判斷,殘差散點分布均勻,說明模型的性能良好[30,31];由圖7的預測殘差可見,各點殘差值即預測響應值和實際響應值之差在0 mg/L上下呈對稱分布,且具有恒定均勻的擴散,表明模型穩定,適于含量預測。因此,本研究成功建立了木犀草苷定量校正模型,并實現了對預測集樣品含量的預測,結果良好,模型穩健有效;在復雜光譜背景下,經過CWT法對樣品的近紅外光譜進行處理和有效檢測信息提取,可以準確、選擇性地測定雪菊提取液中的木犀草苷含量。
4?結 論
采用溫和方法制備了苯硼酸吸附劑,通過紅外光譜進行了表征。將此吸附劑用于雪菊中木犀草苷的富集,與近紅外光譜檢測方法結合,實現了木犀草苷的定量測定。用苯硼酸吸附劑常溫振蕩吸附20 min,對木犀草苷的吸附效率可達到85.5%,吸附效果良好。將其用于雪菊提取液中木犀草苷的富集,采用近紅外漫反射光譜方法對其中微量的木犀草苷進行定量分析,選擇CWT對近紅外光譜進行預處理,可達到最好的交叉驗證結果。用PLSR法建立木犀草苷的定量校正模型,以29個樣本作為預測集進行外部檢驗回歸模型的準確性。在0.15~19.5 mg/L濃度范圍內,預測集相關系數為0.9765,預測集中預測集樣本的回收率在84.7%~113.2%范圍內。結果表明,苯硼酸吸附劑提高了近紅外光譜對木犀草苷的檢測靈敏度,實現了雪菊提取溶液中微量木犀草苷的選擇性檢測,表明基于吸附富集策略與近紅外光譜結合,采用化學計量學方法,可以實現藥食兩用植物中微量成分的分析和測定。
References
1?GUAN Ren-Wei,QU Yong-Sheng,GU Zheng-Wei,SHAO Xin,LIN Hui-Bin,LIN Jian-Qiang. Chinese Wild Plant Resources,2014,33(1): 1-3
管仁偉,曲永勝,顧正位,邵 新,林慧彬,林建強. 中國野生植物資源,2014,33(1): 1-3
2?Li Q L,Tian Z X,Wang M H,Kou J J,Wang C L,Rong X L,Li J,Xie X M,Pang X B. Int. Immunopharmacol.,2019,66: 309-316
3?Wang X Y,Yuan T,Yin N N,Ma X F,Zhang Z B,Zhu Z,Shaukat A,Deng G Z. Inflammation,2018,41(5): 1702-1716
4?Qing W X,Wang Y,Li H,Ma F Y,Zhu J H,Liu X H. AAPS Pharm. Sci. Tech.,2017,18(6): 2095-2101
5?ZHANG Gui-Lin,LIU Min,GE Hong-Juan,SONG Chun-Mei,WANG Dan,ZHANG Ping. Food Science and Technology,2018,43(6): 242-245
張貴林,劉 敏,葛紅娟,宋春梅,王 丹,張 平. ?食品科技,2018,43(6): 242-245
6?Zhang B,Nan T G,Xin J,Zhan Z L,Kang L P,Yuan Y,Wang B M,Huang L Q. J. Pharmaceut. Biomed.,2019,170: 83-88
7?Yang D Z,An Y Q,Jiang X L,Tang D Q,Gao Y Y,Zhao H T,Wu X W. Talanta,2011,85: 885-890
8?Zhou W,Tam K Y,Meng M X,Shan J J,Wang S C,Ju W Z,Cai B C,Di L Q. J. Chromatogr. A,2015,1376: 84-97
9?Feng S X,Li X H,Wang M M,Hao R,Li M M,Zhang L,Wang Z. J. Pharmaceut. Biomed.,2018,148: 205-213
10?Huang Y,Feng Y,Tang G Y,Li M Y,Zhang T T,Fillet M,Crommen J,Jiang Z J. J. Pharmaceut. Biomed.,2017,140: 384-391
11?WANG Shi-Fang,CUI Guang-Lu,FENG Xiao-Yuan,HAN Ping. Journal of Food Safety & Quality,2017,(12): 4602-4608
王世芳,崔廣祿,馮曉元,韓 平. ?食品安全質量檢測學報,2017,(12): 4602-4608
12?GUO Yu-Fei,SU Yi,XU Qing-Yang. Letters in Biotechnology,2016,27(3): 391-395
郭宇飛,蘇 毅,徐慶陽. ?生物技術通訊,2016,27(3): 391-395
13?Xie Y,Zhou R R,Xie H L,Yu Y,Zhang S H,Zhao C X,Huang J H,Huang L Q. Int. J. Biol. Macromol.,2019,122: 1115-1119
14?Chen X Y,Sun X F,Hua H M,Yi Y,Li H L,Chen C. Spectrochim. Acta A,2019,221: 117169
15?YU Hui-Ling,ZHANG Miao,HOU Hong-Yi,ZHANG Yi-Zhuo. Spectroscopy and Spectral Analysis,2019,39(8): 2618-2623
于慧伶,張 淼,侯弘毅,張怡卓. 光譜學與光譜分析,2019,39(8): 2618-2623
