大鼠頸動脈球囊損傷模型內膜增生過程中連接蛋白的變化
2020-05-21 03:44:44潘樂門陳帆風蘇翔楊法鏡
浙江臨床醫學 2020年4期
潘樂門 陳帆風 蘇翔 楊法鏡
血管內皮損傷可誘導動脈內膜增生,引起經皮動脈腔內治療術后的動脈再狹窄。當血管內皮損傷時,血管平滑肌細胞可從中膜層向內膜層遷移,并增殖、分泌細胞外基質,形成再狹窄性病變[1]。經皮腔內治療是針對動脈硬化狹窄疾病的重要治療方法,而腔內治療面臨的主要問題在于如何保持術后的長期通暢率。據估計,近25%的動脈狹窄患者會在血管腔內術后1年內發生再狹窄。內膜增生導致的再狹窄問題,是影響動脈腔內治療術后長期通暢率的重要因素[2]。因此,了解內膜增生過程的機制對于控制術后再狹窄,改善腔內手術效果非常重要。血管內皮細胞與血管平滑肌細胞之間的縫隙連接等細胞間通訊對血管壁的功能至關重要。在動脈粥樣硬化的發展過程中,在血管內皮細胞與血管平滑肌細胞之間的接觸點可以觀察到連接蛋白(connexin,Cx)表達的逐漸變化,并且在動脈粥樣硬化斑塊中可以檢測到連接蛋白表達模式的改變[3]。表明連接蛋白參與動脈粥樣硬化的發展,這種分子是否參與球囊損傷引起的內膜增生和再狹窄的報道較少。在本研究中,通過球囊損傷后再狹窄模型來研究連接蛋白在內膜增生過程中的作用,并初步探索其相關機制。
1 資料與方法
1.1 模型建立 雄性SD大鼠(250±10)g購自北京生命之河實驗動物有限公司,以標準的12h明暗循環進行飼養。將48只大鼠隨機分為對照組(BI)和非損傷組(NI),每組各24只。對照組(BI)采用異氟醚麻醉大鼠,暴露左側頸總動脈,用近端和遠端動脈夾暫時阻斷。將1.5mm球囊導管插入頸動脈并充氣至6atm,然后在短距離內來回拉動3次,以產生內皮損傷。非損傷組(NI)以假手術的大鼠為對照。皮下注射止痛藥和抗生素,恢復期常規監測大鼠。
1.2 組織病理學評價 術后用4%多聚甲醛固定頸動脈,石蠟包埋,沿損傷段行連續切片(2μm),蘇木精-伊紅(H&E)染色檢測內膜增生,利用NIH圖像處理系統獲得內膜、中膜和管腔面積的定量。頸動脈切片進行增殖細胞核抗原(PCNA)染色。用微波加熱提取抗原,4%正常山羊血清加1%牛血清白蛋白(BSA)在PBS中進行非特異性阻斷30min,然后用抗大鼠PCNA兔多克隆抗體(1∶100)孵育切片。二抗選用二氨基聯苯胺底物復合物酶標染色進行信號檢測。最后,用蘇木精對載玻片進行復染和分析。
1.3 Western blot 頸動脈組織加入電泳緩沖液勻化,再經電泳分離,轉膜固定后,加入1∶1000稀釋的Cx30、Cx37、Cx40、Cx43、ERK、Akt、(p)-ERK 和(p)-Akt的抗體在4℃孵育過夜,然后在室溫下加入二抗中進一步孵育1h。用Image J軟件計算磷酸化與總蛋白表達的比率。
1.4 數據處理 所有實驗均用GraphPad Prism 6進行定量分析,結果以()表示,并采用t檢驗或單因素方差分析,隨后進行Tukey后檢驗。P<0.05為差異具有統計學意義。
2 結果
2.1 連接蛋白的時間表達 在整個研究期間,BI和NI大鼠術后均保持健康,體重無明顯下降。兩組分別于術后6h、24h、7d、14d處死6只大鼠,進行Cx30、Cx37、Cx40和Cx43的檢測。Western blot結果表明,隨著時間的推移,Cx37表達呈下降趨勢,Cx43表達呈上升趨勢,而Cx30和Cx40表達無顯著差異(見圖1)。結果表明,在四種連接蛋白中,只有Cx37和Cx43在球囊損傷后的整個過程中表達有明顯差異。
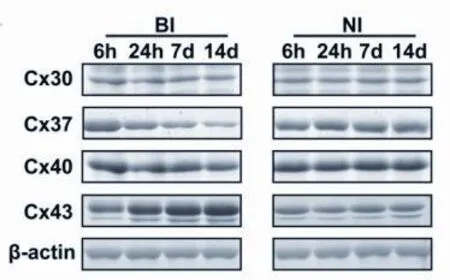
圖1 連接蛋白的時間表達情況。為Cx30、Cx37、Cx40和Cx43這四種連接蛋白在術后6h、24h、7d和14d等4個時間點的Western印跡分析
2.2 球囊損傷致新生內膜形成 根據連接蛋白表達的結果,通過HE染色觀察術后第14天球囊損傷對新生內膜形成的影響(每組6只)。在接受假手術的大鼠中未觀察到明顯的內膜增生,而在BI組中發現顯著的內膜增生(見圖2A)。定量形態計量學評估顯示,BI組的內膜面積(見圖2B,P<0.01)和內膜-中膜(I/M)比值(見圖2E,P<0.01)較NI組顯著增加,且BI組的管腔面積明顯低于NI組(見圖2D,P<0.01),而兩組之間的內外膜間的區域無顯著差異(見圖2C)。
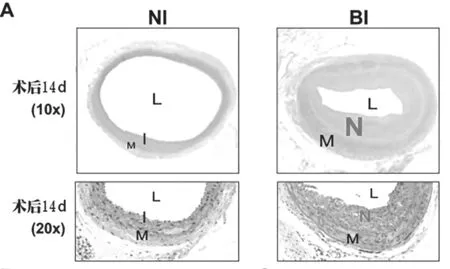
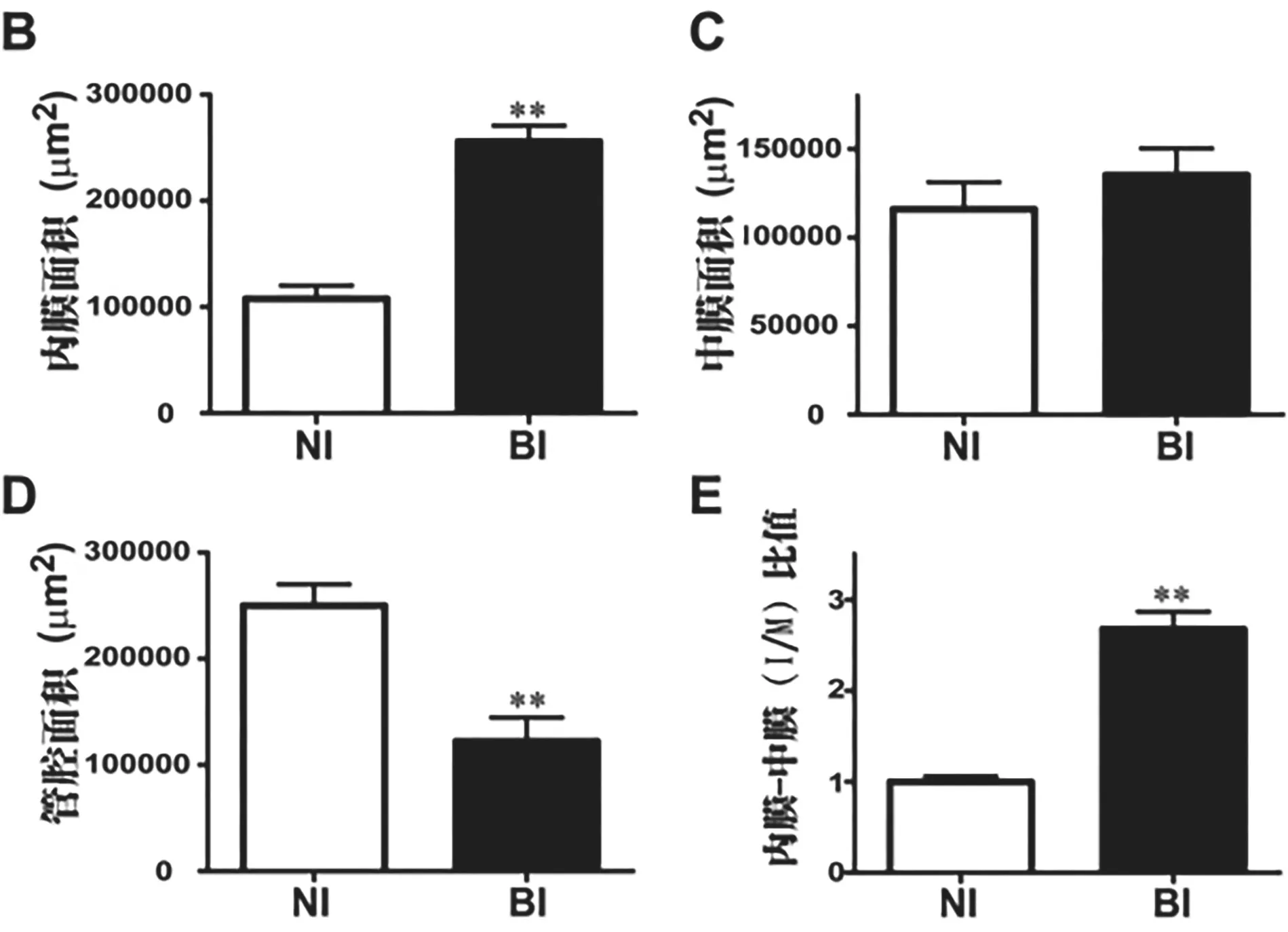
圖2 球囊損傷促進新生內膜形成情況。(A)頸總動脈橫截面的典型H&E染色顯微照片。L:管腔;M:中膜;I:內膜;N:新生內膜。(B)內膜面積的定量分析。(C)中膜面積的定量分析。(D)管腔面積的定量分析。(E)內膜與中膜(I/M)比值的定量。★★P<0.01
2.3 ERK和Akt表達上調 ERK途徑和PI3K/Akt途徑被證實是新生內膜形成的關鍵。在本研究中,總ERK、p-ERK、總Akt和p-Akt在BI大鼠中呈上調表達(見圖3A),與NI大鼠相比,Akt和ERK的磷酸化比率也呈上調表達(見圖3B,P<0.01)。由此推測,Cx37和Cx43的表達可能通過ERK和Akt通路調節。
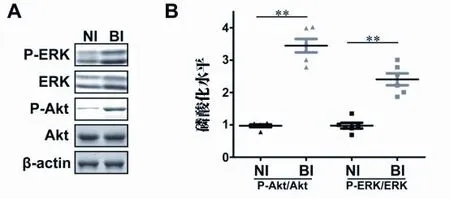
圖3 ERK和Akt的磷酸化水平。(A)Akt和ERK磷酸化水平的Western印跡分析,以β-肌動蛋白為對照。(B)Akt和ERK磷酸化水平的量化。通過比較Akt和ERK的磷酸化比率,可以發現球囊損傷組磷酸化水平顯著升高。★★P<0.01。
3 討論
連接蛋白是縫隙連接的組成蛋白,而縫隙連接是一簇連接兩個細胞的通道,由連接蛋白體和半通道組成,分別來自于相連接的兩個細胞。這種細胞間的通道能夠促進化學和電信號的傳播,對于血管的功能具有重要作用[4]。在21種已經鑒定的連接蛋白中,血管內皮細胞主要表達Cx37、Cx40和Cx43[5]。
在本研究中,Cx37和Cx43均參與了球囊損傷引起的內膜增生,其機制可能與ERK和Akt通路的調節相關。實驗證明,機械損傷誘導的生長因子釋放可激活ERK級聯,使縫隙連接蛋白能參與組裝、分解、運輸和降解的調節,以及縫隙連接通道的門控[6]。而Akt在內膜增生過程中的作用則較少被討論。本研究提示ERK和Akt均參與調解內膜增生,但具體機制尚需進一步研究。
連接蛋白在不同組織中的不同表達模式表明,通過縫隙連接可能有分子運動特異性。值得注意的是,Cx40/Cx43的表達率可以影響異質/異型縫隙連接通道的特性。此外,連接蛋白的磷酸化可以調節連接通透性[7]。Cx37和Cx43的共表達、相互表達或磷酸化調節是否參與球囊損傷誘導的內膜增生尚需進一步研究。
靜止的血管平滑肌細胞重新進入細胞周期,進行增殖和遷移是內膜增生過程中的標志性生物學事件,通常認為這是由血管平滑肌細胞與內皮細胞之間的縫隙連接通訊所調控的。因此,有必要深化對連接蛋白調控的研究。
雖然球囊損傷誘導內膜增生的具體機制尚不清楚,但本研究提示ERK和Akt介導的下調Cx37和上調Cx43參與了這一過程。總之,球囊損傷誘導的內膜增生過程中,連接蛋白Cx37下調,Cx43上調,其機制可能與ERK和Akt的介導相關。
