取代水楊醛席夫堿金屬配合物結構對硫化物催化氧化性能的影響
2020-05-27 01:13:00姜翠玉梁書源賈超洋張龍力
石油學報(石油加工) 2020年3期
姜翠玉, 梁書源, 賈超洋, 劉 蕾, 邵 雪, 張龍力
(1.中國石油大學(華東) 理學院,山東 青島 266580;2.中國石油大學(華東) 化學工程學院,山東 青島 266580)
硫和含硫化合物廣泛存在于石油產品中,如何降低油品的硫含量是儲運、加工和使用這些油品時面臨的一項巨大挑戰。燃料油脫硫方式主要有堿洗脫硫[1]、加氫脫硫[2-4]和氧化脫硫[5-7]。傳統的堿洗脫硫方法僅能脫除部分酸性硫化物,如硫化氫和小分子硫醇等,但會產生大量有害硫酸鹽廢液和廢堿液,易造成環境污染;加氫脫硫方法盡管脫硫效率較高,但很難脫除雜環硫化物,反應條件苛刻,且成本高、投資大。相比之下,氧化脫硫的脫硫效率較高、條件更溫和[8]、成本更低、適用性更強。但目前的氧化脫硫工藝中,氧化劑常用過氧化氫和有機過氧化物,消耗量巨大且存在嚴重的安全隱患[9]。
席夫堿金屬配合物是一種高效的催化氧化反應催化劑[10-11],已被用于苯甲醇[12]、環己烷[13-14]等化合物的催化氧化,但迄今未見用于油品氧化脫硫的研究報道。如果席夫堿金屬配合物對油品中的硫化物的脫除具有明顯的催化效果,則可能發展成為一個新的研究領域,并有可能影響傳統的脫硫技術。
筆者以具有不同電子效應和位阻效應取代基的水楊醛為原料,分別與乙二胺和鄰苯二胺反應合成7種席夫堿配體,然后將合成的配體分別與硝酸鈷和硝酸鎳反應,制備7種席夫堿金屬配合物(C1~C7),并考察了其摩爾載氧量與溶解度。然后以合成的配合物作為催化劑,以氧氣為氧化劑,考察其對含1-己硫醇、二丁基硫醚和2-甲基噻吩的模型硫化物體系的催化氧化性能,探討影響席夫堿金屬配合物催化氧化脫硫活性的因素,并從中心金屬離子種類、配體的電子效應和空間效應以及配合物溶解度等方面進行配合物構效關系探究,為進一步開展席夫堿金屬配合物在燃料油催化氧化脫硫方面的研究和應用打下一定的基礎。
1 實驗部分
1.1 試劑與儀器
試劑:乙醇、硝酸鈷、硝酸鎳、正辛烷、氫氧化鈉,均為分析純,國藥集團化學試劑有限公司產品;乙二胺、鄰苯二胺、水楊醛、鄰香蘭素、5-氯水楊醛、3,5-二叔丁基水楊醛、1-己硫醇、二丁基硫醚、2-甲基噻吩、N,N-二甲基甲酰胺,均為分析純,阿拉丁試劑公司產品。
儀器:RE-52A型旋轉蒸發儀,上海亞榮生化儀器廠產品;7820A型氣相色譜,安捷倫(上海)科技有限公司產品。
1.2 席夫堿金屬配合物的合成
按照反應式(1)所示,分別將水楊醛、鄰香蘭素、5-氯水楊醛、3,5-二叔丁基水楊醛與乙二胺按n(醛)∶n(胺)=2∶1比例制備席夫堿配體L1~L4;按照反應式(2)所示,將水楊醛、3,5-二叔丁基水楊醛與鄰苯二胺按n(醛)∶n(胺)=2∶1比例制備席夫堿配體L5和L6。然后,將L1按照反應式(3)分別與Co(NO3)2、Ni(NO3)2反應合成配合物C1和C2;將L2~L4按反應式(3)與Co(NO3)2反應合成配合物C3~C5;將L5、L6按式(4)與Co(NO3)2反應合成配合物C6、C7。具體合成步驟參照文獻[15]進行。
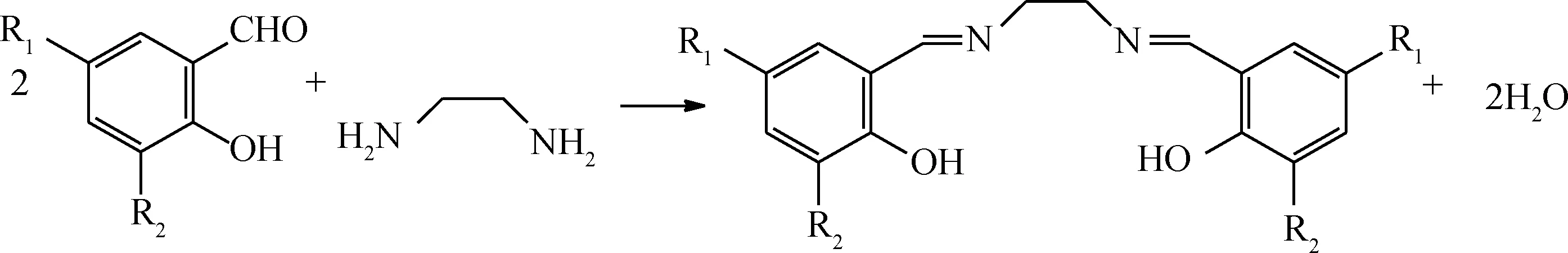
(1)
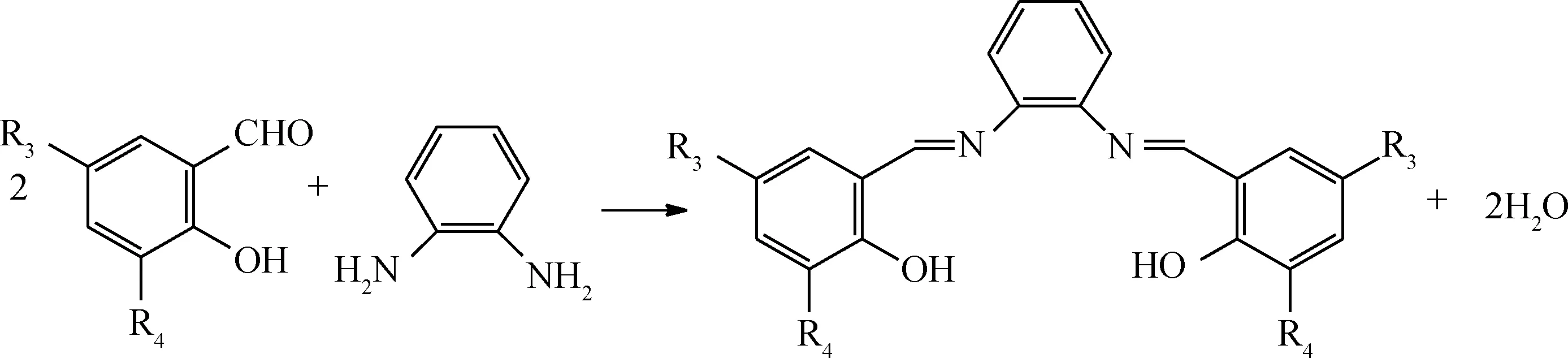
(2)
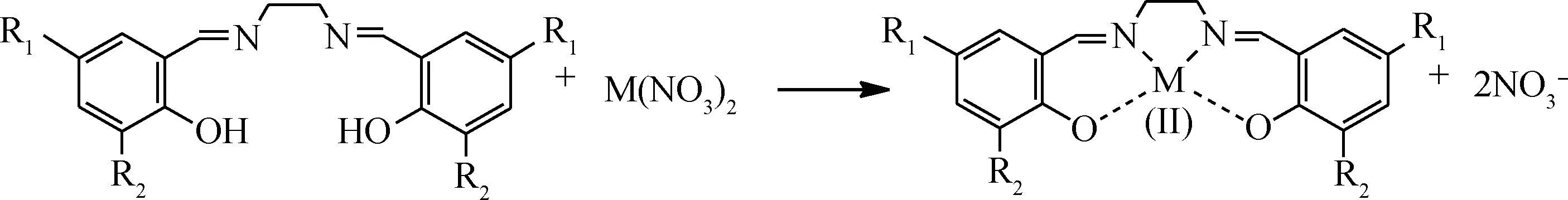
(3)
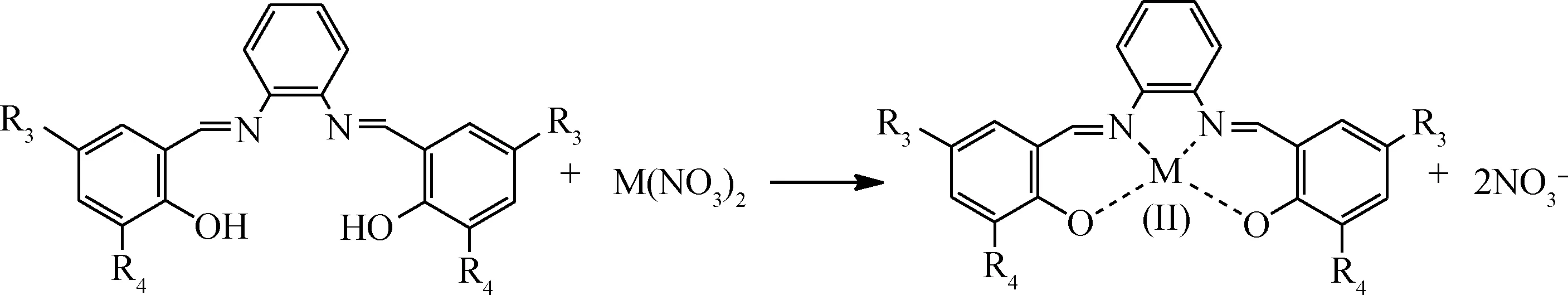
(4)
1.3 配合物載氧量及溶解度測定
1.3.1 配合物載氧量測定
根據文獻[16]設計載氧實驗裝置,如圖1所示。以C1為例,稱取0.3 g配合物C1置于圓底燒瓶中,在恒壓漏斗中加入30.0 mL的DMF(N,N-二甲基甲酰胺)作溶劑;向量氣管中注入一定量的水,記錄水面初始高度,保證量氣管垂直;向實驗體系通入O2,檢驗并確保實驗裝置的密封性。記錄室溫T以及大氣壓p。將DMF加入燒瓶,開啟攪拌并計時。每隔2 min記錄量氣管液面變化,直至液面恒定。繪制載氧體積V隨時間t變化的曲線,得到0.3 g配合物最大載氧量后,由式(5)計算出每摩爾配合物吸收的O2的物質的量。
(5)
式(5)中,n為摩爾載氧量,即每摩爾配合物吸收O2的物質的量,mol/mol;m為配合物質量,g;M為配合物分子相對分子質量,g/mol;p為實驗進行時的大氣壓,Pa;V為吸收O2的體積,mL;R為理想氣體常數,J/(mol·K);T為實驗進行時的室溫,K。
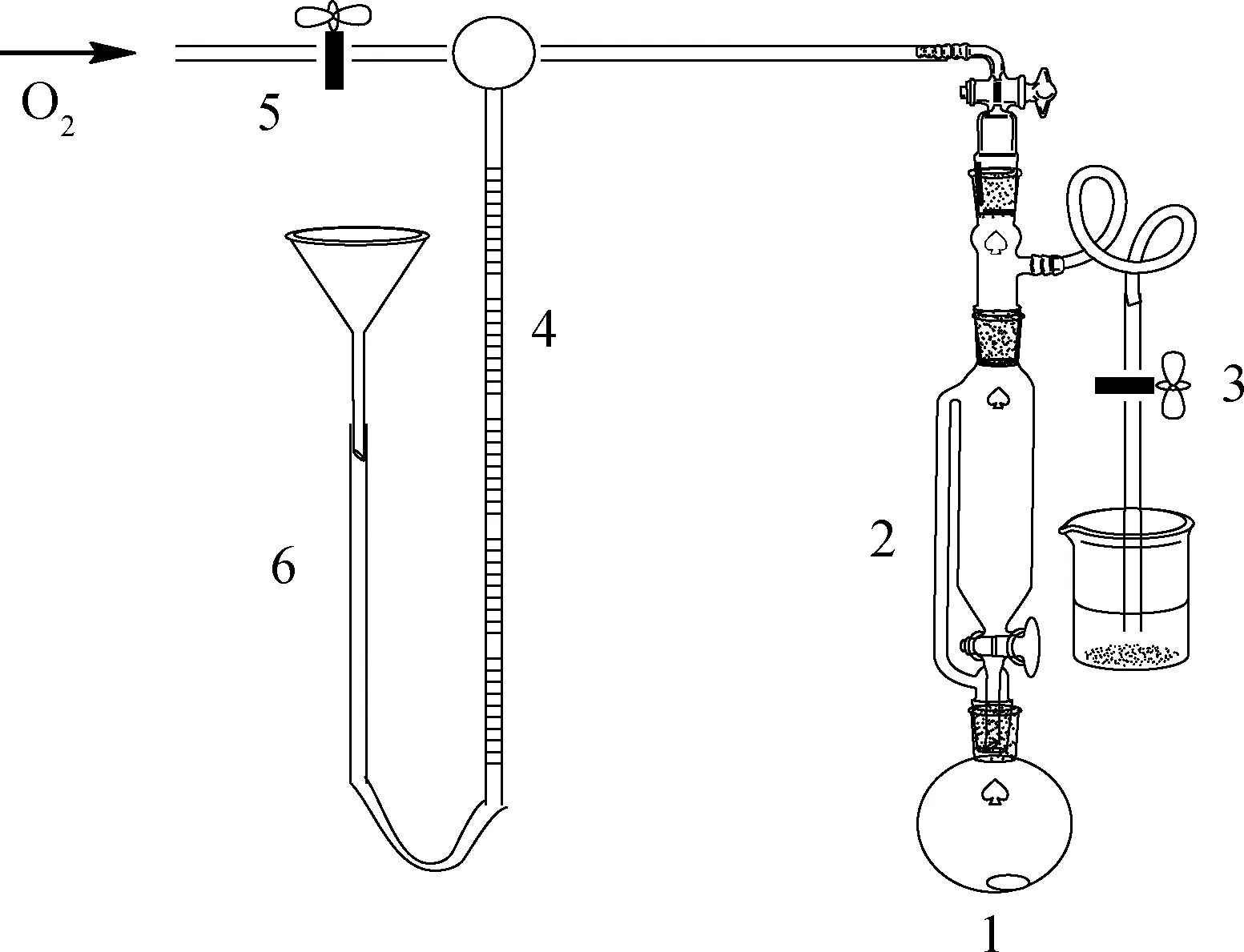
圖1 載氧實驗裝置Fig.1 Oxygen carrying experimental device1—Round-bottomed flask; 2—Constant pressure funnel;3,5—Cock; 4—Trachea; 6—Glass tube
1.3.2 配合物在DMF及正辛烷中溶解度測定
由于DMF是載氧實驗所用溶劑,而正辛烷是模擬油體系的溶劑,配合物在二者中的溶解度會影響載氧性能和反應性能。故采用靜態平衡法[17]測定C1~C7在DMF和正辛烷中的溶解度。方法:將配合物分別溶解于DMF和正辛烷,得到飽和溶液;稱量空圓底燒瓶質量m2,取10.0 mL配合物飽和溶液于燒瓶中,蒸干溶劑,烘干至恒重,稱重得m1,配合物溶解度S根據式(6)計算。
S=(m1-m2)/10
(6)
式(6)中,m1為蒸干溶劑后剩余溶質和圓底燒瓶的質量,g;m2為圓底燒瓶質量,g;S為配合物溶解度,g/mL。
1.4 模型硫化物的催化氧化性能考察
1.4.1 模擬油催化氧化實驗
將1.0 mL 1-己硫醇、二丁基硫醚、2-甲基噻吩分別加入 200.0 mL 正辛烷中,配制成模擬油。在帶有磁力攪拌、冷凝管的250 mL三口燒瓶中依次加入 0.10 g 配合物、100.0 mL模擬油和30 mL的NaOH水溶液(質量分數為30.0%)。水浴溫度調節至80 ℃,攪拌轉速1200 r/min,使得水溶液與模擬油層充分混合,以0.80 L/min的氣速向體系中通入氧氣進行反應。考察氧化時間為15、30、45、60、75 min時,配合物對模擬油中硫化物的催化氧化效果。
1.4.2 模型硫化物氧化轉化率測定
1-己硫醇、二丁基硫醚、2-甲基噻吩在配合物C1~C7催化下,被氧氣氧化后皆生成相應的砜類或亞砜類物質。其中1-己硫醇、二丁基硫醚還進一步被氧化生成SO32-或SO42-。按照文獻[18]采用氣相色譜分析氧化前、后體系中3種模型硫化物的含量,計算其氧化轉化率。分析前,在油樣中加入1.0 μg的1-己硫醇、二丁基硫醚和2-甲基噻吩標準物質,根據標準物加入前、后峰面積的變化,確定各硫化物含量。氣相色譜分析條件為:HP-5色譜柱(30.0 m×0.25 mm×0.25 μm);FID檢測器溫度為300 ℃;采用程序升溫法升溫;分流比為10∶1;載氣為氮氣,流量為20 mL/min。
為了避免硫化物的揮發引起的測量誤差,氧化t分鐘后油樣中硫化物a的質量mat計算式如式(7)所示。
(7)
式(7)中,A和A1分別為標準物質加入前和加入后硫化物a的峰面積。氧化t分鐘后硫化物a的氧化轉化率ηat計算公式如式(8)所示。
(8)
式(8)中,ma和mat分別為氧化前、后油樣中硫化物a的質量,mg。
2 結果與討論
2.1 配合物的載氧性能及溶解度
2.1.1 配合物的載氧性能
7種配合物的摩爾載氧量和達到最大載氧量的時間如表1所示,其中n為摩爾載氧量,tmax為達到最大載氧量所需要的時間。由表1可知,7種配合物的摩爾載氧量由高到低順序為C7、C6、C3、C1、C2、C5、C4,每摩爾C7配合物最多可以吸收0.523 mol O2。席夫堿配合物的載氧性能主要由其結構決定:中心離子與氧分子的配位能力越強、配體取代基推電子能力越大且空間位阻越小、共軛鏈越長,越有利于載氧。如:由鄰苯二胺制得的配合物C7、C6的載氧能力明顯高于以乙二胺合成的配合物C1~C5,因為鄰苯二胺分子的共軛程度更大,配合物與O2結合形成的中間體結構更穩定,其載氧能力也更好;而C1與C2相比,席夫堿配體相同,中心金屬離子分別為Co(Ⅱ)和Ni(Ⅱ)離子,Co(Ⅱ)離子的電子密度高于Ni(Ⅱ)離子,因此C1吸附氧的能力優于C2,載氧能力更強;對于C3、C1、C5和C4,其載氧能力因配體苯環上的取代基的不同而不同。C3中苯環3號位含有強推電子基團-OCH3,使苯環電子云密度增大并通過共軛效應增強了中心金屬離子的電子云密度,因此其載氧能力最大。C4苯環含吸電子基團-Cl,降低了中心金屬離子的電子云密度,因此其載氧性能較差。C5盡管含有推電子基團-C(CH3)3,但由于2個叔丁基尤其是3號位的叔丁基存在較大的空間位阻效應,影響配合物的共平面性從而影響其共軛的程度,造成其載氧性能較低。
此外,7種配合物達到最大載氧量的速率由大至小順序為C4、C5(C3)、C6、C1、C7、C2。其中,C5與C3載氧速率相同。
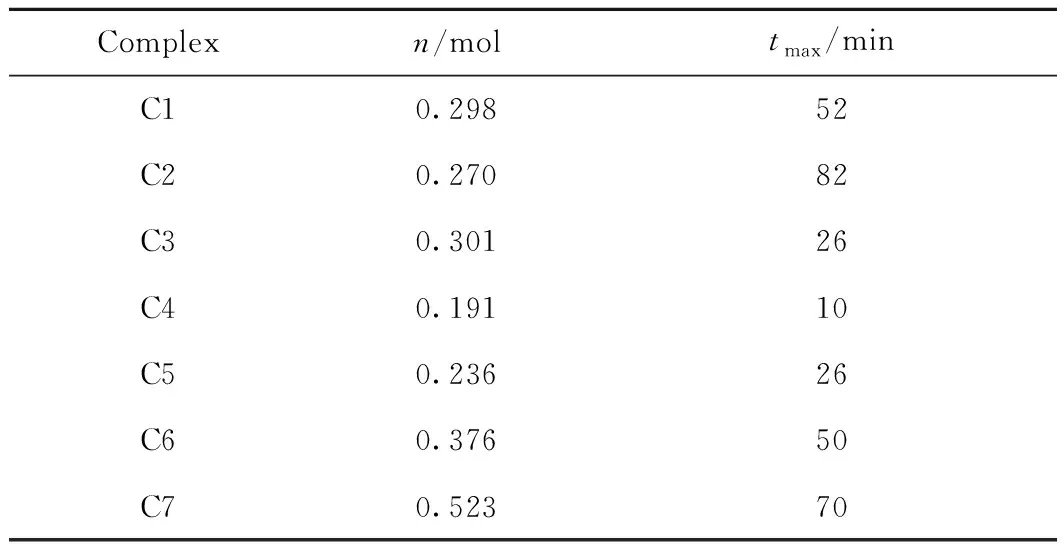
表1 C1~C7摩爾載氧量與達到最大載氧量的時間Table 1 Molar oxygen carrying property and the time to reach maximum oxygen carrying of C1-C7
2.1.2 配合物在不同溶劑中的溶解度
室溫下,配合物C1~C7在DMF和正辛烷中的溶解度如表2所示。由表2可知,C1~C7在DMF中都能較好地溶解;而在正辛烷中溶解度較小且差別較大。其在正辛烷中的溶解度由大到小順序是C5、C6、C7、C2、C1、C4、C3。分析原因,C5苯環上帶有2個疏水性-C(CH3)3基團,極性最小,在正辛烷中的溶解度最高;C6和C7中含有更多的疏水性芳香環,溶解度也較高;而C3因苯環上的-OCH3有一定親水性,使其在正辛烷中的溶解度最低。在正辛烷中溶解度越大,意味著配合物在溶劑中分散得越均勻,有效濃度越高,氧化過程中的傳質效果更好。配合物不溶解的部分是以固體顆粒的形式漂浮于油-水界面或者沉降至底部,對氧化過程中的傳質作用有較大的不利影響。
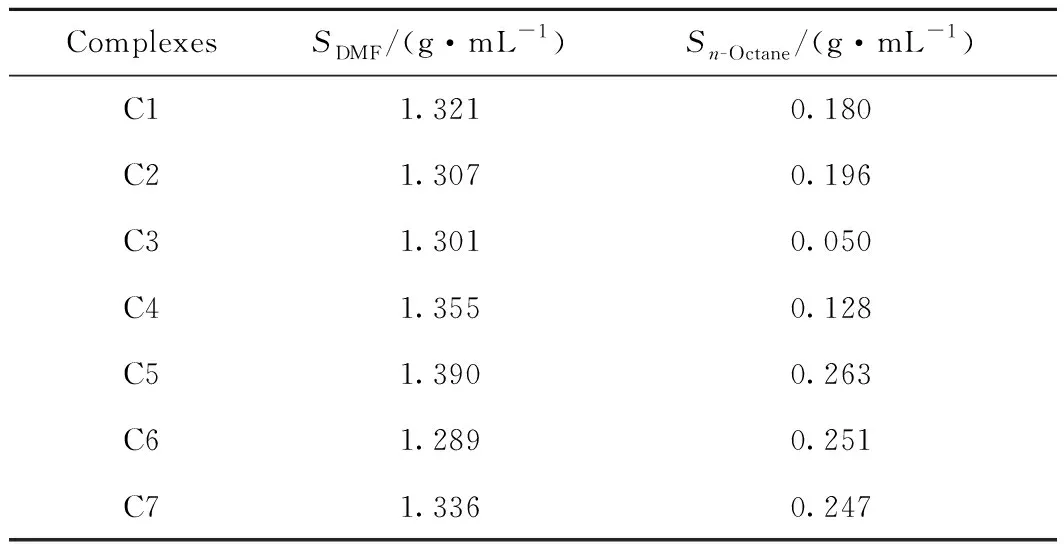
表2 配合物C1~C7在DMF以及正辛烷中的溶解度Table 2 The solubility of C1-C7 in DMF/ n-octane
2.2 配合物的催化氧化性能考察
對模擬油進行催化氧化實驗發現,由于NaOH水溶液對硫醇具有堿洗脫除作用,1-己硫醇在 10 min 左右就接近100%的轉化率[15]。為排除NaOH水溶液堿洗作用對硫醇等脫除率的影響,催化氧化反應在非堿性體系中進行。反應75 min內,模擬油中1-己硫醇、二丁基硫醚和2-甲基噻吩的氧化轉化率隨氧化時間的變化曲線如圖2所示。其中,圖2(a)~(g)分別對應配合物C1~C7催化的氧化脫硫反應。
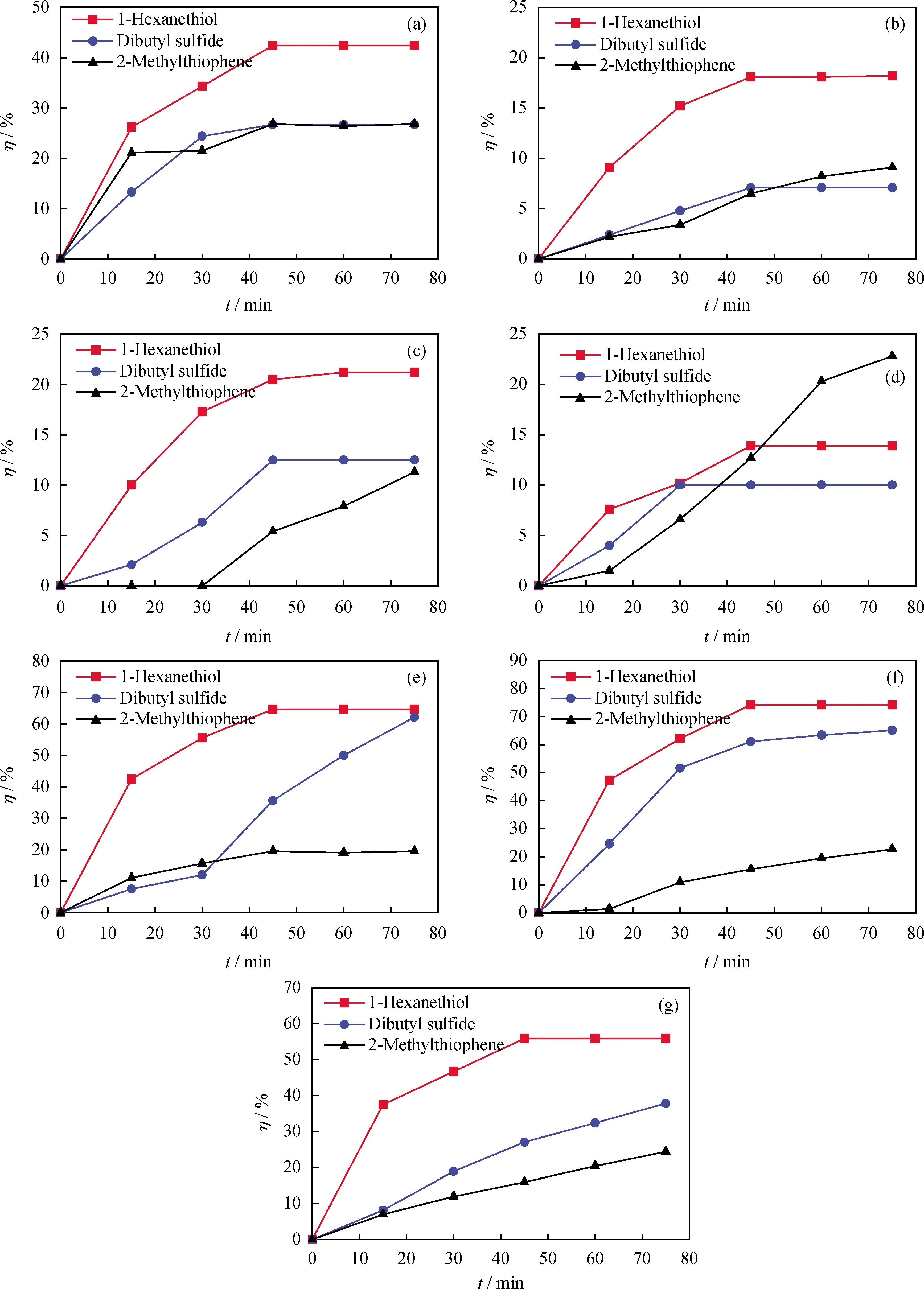
圖2 不同配合物催化下各硫化物氧化轉化率與時間的關系Fig.2 Relationship between conversion rate and time of various sulfides catalyzed by different complexes(a) C1; (b) C2; (c) C3; (d) C4; (e) C5; (f) C6; (g) C7Reaction conditions: m(Catalyst)=0.1 g; F(O2)=0.8 L/min; V(Oil)=100.0 mL
由圖2可知:隨著反應時間的延長,3種硫化物在不同配合物催化下的氧化轉化率基本上都呈先上升后平穩的趨勢;C4(圖2(d))對三者的氧化性能都較弱;除以C4為催化劑的反應外,其他催化氧化反應中1-己硫醇的氧化轉化率均高于二丁基硫醚和2-甲基噻吩;C5(圖2(e))、C6(圖2(f))催化二丁基硫醚的氧化轉化率明顯高于2-甲基噻吩,趨近于1-己硫醇;配合物C6對3種硫化物的催化氧化轉化率都較高,1-己硫醇的氧化轉化率最高可達74.2%,二丁基硫醚的氧化轉化率為65.1%,而7種配合物對2-甲基噻吩的催化氧化效果都較差,主要是因為2-甲基噻吩環系較穩定,且噻吩硫上的電子云密度較低[19],不易被氧化。
2.3 影響配合物催化氧化性能的因素
2.3.1 中心金屬離子對配合物催化性能的影響
配體相同,中心金屬離子分別為Co(Ⅱ)和Ni(Ⅱ)離子的配合物C1和C2催化氧化3種硫化物的轉化率比較如圖3所示。由圖3可知,C1對3種硫化物的催化氧化性能均優于C2。原因在于,C1中心金屬離子Co(Ⅱ)更易與氧氣形成配合物,使C1吸附、活化分子氧的能力和穩定性都好于C2[13],即C1載氧性能優于C2。同時,二者在正辛烷中的溶解度相近,說明其催化氧化性能主要受其載氧性能影響。因此,配體相同情況下,中心金屬離子為Co(Ⅱ)的配合物的催化性能優于中心離子為Ni(Ⅱ)的配合物。
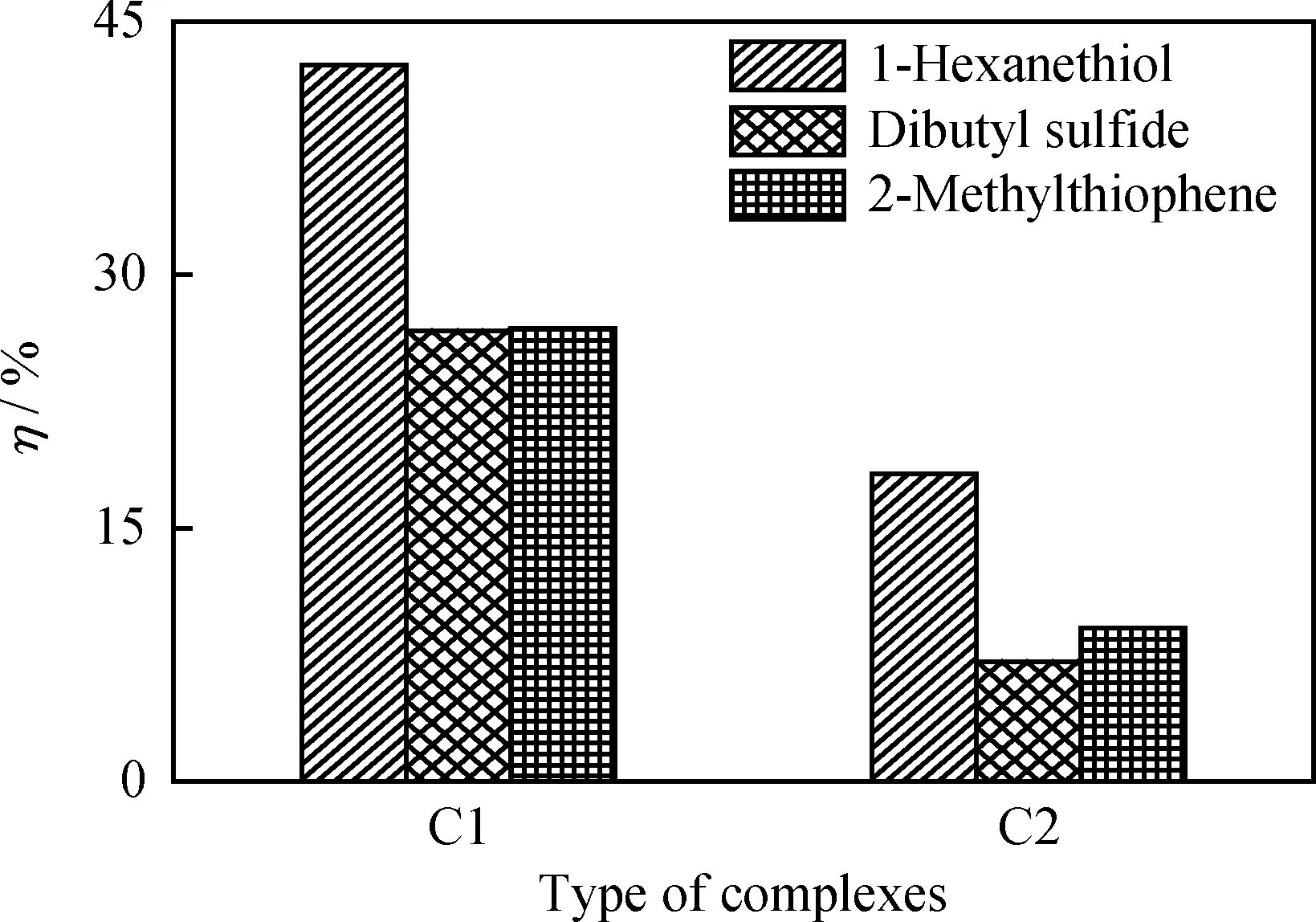
圖3 中心離子對配合物催化氧化性能的影響Fig.3 Effects of the central ion on catalytic oxidation property of complexesReaction conditions: m(Catalyst)=0.1 g; F(O2)=0.8 L/min;V(Oil)=100.0 mL; t=75 min
2.3.2 水楊醛苯環上的取代基對配合物催化性能的影響
考察C1、C3、C4、C5對模型硫化物的催化氧化性能,結果如圖4所示。由圖4可知,配合物催化氧化硫化物性能由高到低的順序為C5、C1、C3、C4。配合物的催化氧化性能與其載氧性能規律并不完全一致,說明還可能受其他因素影響。
配合物C1、C3、C4、C5結構上的主要區別在于水楊醛苯環上取代基的不同:C1中水楊醛苯環上沒有取代基;C3、C5苯環上分別有推電子基 -OCH3和-C(CH3)3,且推電子能力-OCH3大于-C(CH3)3;而C4苯環上連有吸電子基-Cl。圖4 結果表明,苯環上有吸電子基的配合物對硫化物的催化氧化性能最差;苯環上有推電子基的配合物性能較好;但含有強的推電子基-OCH3的配合物C3的催化氧化性能卻低于沒有取代基的C1,這說明配合物的催化氧化性能不僅僅取決于取代基的吸、推電子能力,還受其在溶劑中溶解性能的影響。盡管-OCH3的推電子能力最強,使C3的載氧量最大,但是由于其親水性較強,導致C3在正辛烷中的溶解度最小,從而影響C3對模擬體系中硫化物的催化氧化性能。
因此,配合物對硫化物的催化氧化性能受水楊醛苯環上取代基及其在體系中的溶解度的共同影響。在選擇原料水楊醛時,既要考慮取代基的電子效應又要考慮其溶解性。C5的載氧能力雖然稍差,但是其在正辛烷中的溶解度最大,較高的濃度有利于配合物與硫化物的相互作用,彌補了其載氧性能不佳的劣勢,使其具有相對更好的催化氧化性能。
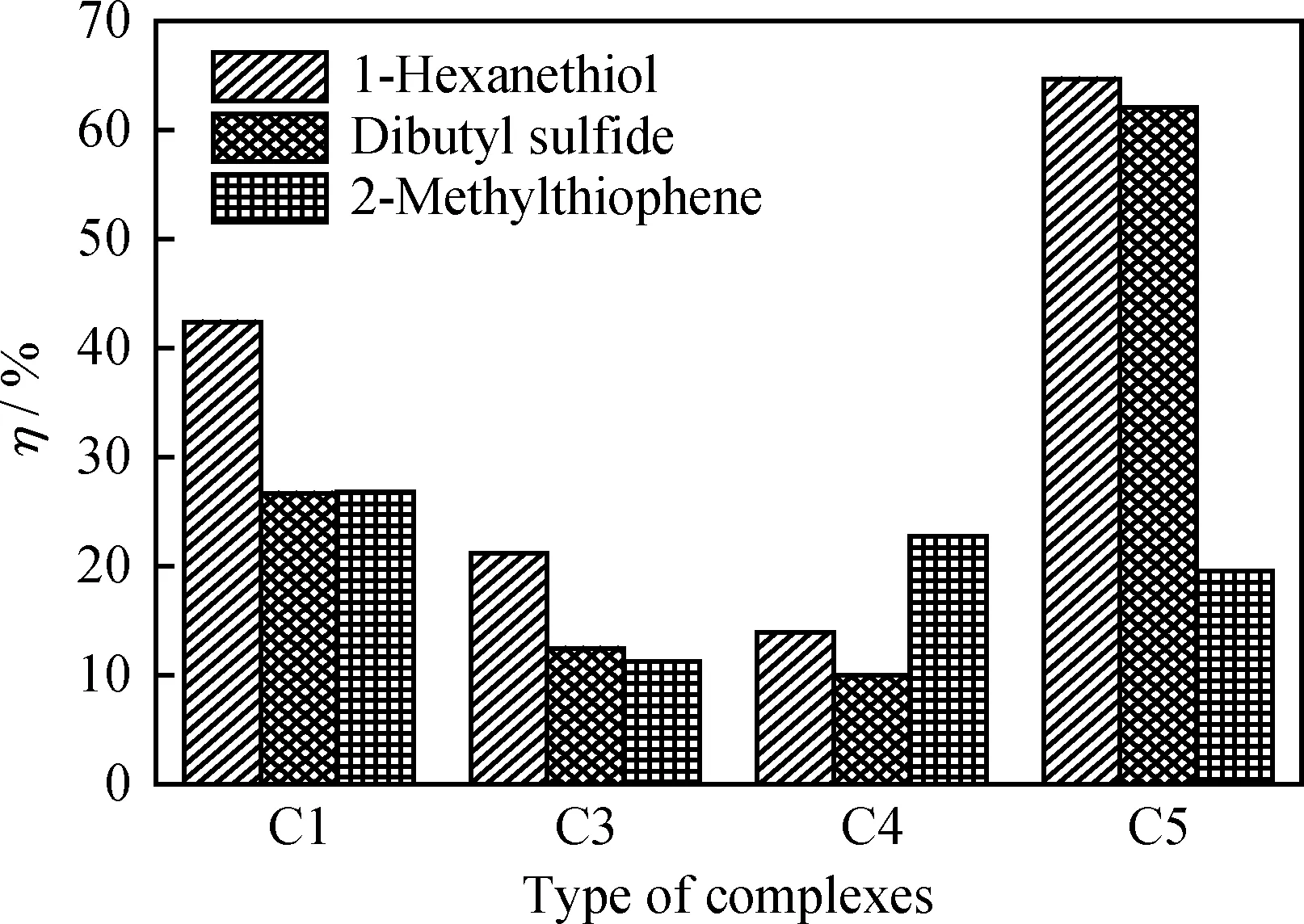
圖4 苯環取代基對配合物催化氧化性能的影響Fig.4 Effects of substituent groups on catalytic oxidation property of complexesReaction conditions: m(Catalyst)=0.1 g; F(O2)=0.8 L/min;V(Oil)=100.0 mL; t=75 min
2.3.3 反應物胺的結構對配合物催化性能的影響
配合物C1和C6對硫化物的催化氧化性能如 圖5 所示。由圖5可知,C6的催化氧化性能比C1更好。分析原因主要是二者合成原料二胺的結構不同所致,C6是由鄰苯二胺制備得到的,其分子的整體共軛程度大于由乙二胺合成得到的C1,配合物C6與O2結合形成的中間體結構更穩定,使其載氧性能和在正辛烷中的溶解度都比C1更大。
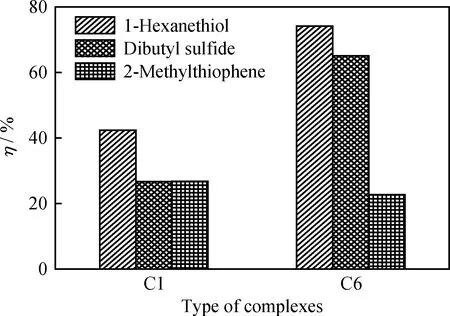
圖5 不同的二胺對配合物催化氧化性能的影響Fig.5 Effects of different diamines on catalytic oxidation property of complexesReaction conditions: m(Catalyst)=0.1 g; F(O2)=0.8 L/min;V(Oil)=100.0 mL; t=75 min
3 結 論
(1)配合物C1~C7在DMF中溶解度較大且幾乎沒有差別;在正辛烷中溶解度差別較大,且由大到小順序是:C5、C6、C7、C2、C1、C4、C3;配合物中含有的疏水性基團越多,其在非極性溶劑正辛烷中的溶解度則越大。
(2)配合物C1~C7的載氧能力由高到低的順序為C7、C6、C3、C1、C2、C5、C4。配合物中心金屬離子電子云密度越大,配體取代基推電子能力越大、空間位阻越小、共軛鏈越長,則其載氧能力越強。
(3)配合物C1~C7催化氧化硫化物的性能受其載氧能力和在正辛烷中的溶解度共同影響,其載氧能力越強,在正辛烷中溶解度越大,則催化氧化硫化物的性能越強。配合物對不同硫化物的催化氧化能力不同。對3種硫化物催化氧化性能效果最好的配合物為C6,其催化氧化1-己硫醇的轉化率達到74.2%,二丁基硫醚的轉化率為65.1%,2-甲基噻吩的轉化率較低。
