QuEChERS-超高效液相色譜-串聯(lián)質譜法快速測定辣椒紅色素中9 種非食用色素
2020-06-01 04:07:48牛瑜琦馬曉斐張敬軒高文惠
食品科學 2020年10期
牛瑜琦,馬曉斐,李 揮,張敬軒,,高文惠,
(1.河北科技大學生物科學與工程學院,河北 石家莊 050018;2.河北冠卓檢測科技股份有限公司,河北 石家莊 051130;3.河北省醫(yī)療器械與藥品包裝材料檢驗研究院,河北 石家莊 050061)
辣椒紅色素是一類存在于成熟的紅辣椒的果皮中的天然植物提取物著色劑,屬于類胡蘿卜素,具有著色力強、色澤鮮艷、保色效果好、無毒無害等優(yōu)點,廣泛用于食品、藥品、飲料、化妝品、農(nóng)副加工產(chǎn)品等,使用范圍和市場需求日益增長,其質量安全備受關注。然而由于環(huán)境污染,工業(yè)染料存在于土壤、空氣等環(huán)境中,在紅辣椒的生長過程中可能會帶入一些非食用色素物質,或者在儲存和運輸過程中污染植物;在辣椒紅色素的制備過程中,伴隨著提取、精制等加工過程而濃縮富集,同時非食用色素在提取過程中不易揮發(fā)、降解,從而導致在辣椒紅色素中含有一定量這類有害物質,并最終轉移至食品中,導致食品安全問題[1-5]。辣椒紅色素基質復雜,檢測干擾大,從環(huán)境可能帶入的非食用色素種類多,含量低。因此,亟待需要高靈敏度、高特異性的檢測技術。
目前,對于非食用色素的檢測方法主要有免疫法[6-7]、電化學分析法[8-9]、薄層色譜法[10-11]、液相色譜法[12-14]、高效液相色譜-質譜法[15-17]等。免疫法不滿足多殘留藥物同時檢測,易出現(xiàn)假陰性或假陽性以應。電化學分析法和薄層色譜法特異性差、靈敏度低、選擇性不高、定性能力較差。液相色譜法靈敏度較低、選擇性差。高效液相色譜-質譜法因具有高的靈敏度、特異性和抗干擾能力,已成為痕量藥物分析的首選方法。陳林等[18]采用改良QuEChERS(quick, easy, cheap, effective, rugged and safe)技術作為前處理方法,建立了辣椒制品中14 種非法添加工業(yè)染料的液相色譜-串聯(lián)質譜聯(lián)用快速檢測方法;路楊等[19]采用凝膠滲透色譜儀作為前處理設備,采用超高效液相色譜儀作為檢測設備,建立辣椒油和火鍋底料中的10 種工業(yè)染料的檢測方法。同時,針對當前天然植物提取物中非食用色素檢測技術缺失,缺乏相關檢測標準,建立高通量超痕量的檢測技術,對檢測方法進行實驗室內(nèi)、間的驗證實驗,考察方法學指標,為食品安全監(jiān)管和溯源提供技術依據(jù)。因此,對植物提取物中非食用色素的檢測方法技術進行研究非常必要。另外,傳統(tǒng)的前處理方法比較耗時費力,而QuEChERS法萃取具有“快速、簡易、廉價、有效、穩(wěn)定、安全”的特點[20-26],該方法是Anastassiades等[27]于2002年首先在歐洲農(nóng)藥殘留研討會提出,并于2003年正式發(fā)表了一種用于水果、蔬菜、谷物等低脂產(chǎn)品中多農(nóng)藥殘留分析的前處理方法。QuEChERS方法具有技術要求低、操作簡單、無需使用大量有機溶劑、分析過程快速等優(yōu)點。
本研究以辣椒紅色素為研究對象,選用QuEChERS前處理法,分別對提取溶劑、脫水劑、吸附劑進行優(yōu)化,建立QuEChERS-超高效液相色譜-串聯(lián)質譜檢測方法,并對前處理基質效應和建立的方法進行評價和驗證。為辣椒制品的食品添加劑中非法添加工業(yè)染料的檢測提供一種更加快速、簡便的技術支持,解決高濃縮提取物基質干擾嚴重的難題,為食品中非食用物質溯源技術提供參考,為食品安全監(jiān)管提供有力的技術支持。
1 材料與方法
1.1 材料與試劑
辣椒紅色素樣品 晨光生物科技集團股份有限公司;乙腈、甲醇、乙酸(均為色譜純) 德國默克公司;乙酸乙酯、正己烷、乙醚(均為分析純) 賽默飛世爾科技有限公司;氯化鈉為分析純;實驗所用水均為超純水(電阻率為18.2 MΩ·cm);蘇丹紅I(100 μg/mL)、蘇丹紅II(100 μg/mL)、蘇丹紅III(100 μg/mL)、蘇丹紅IV(100 μg/mL)、羅丹明B(95.52%)、堿性橙II(99.6%)、堿性橙21(98%)、堿性橙22(99.9%)、堿性嫩黃(79.5%)標準物質 德國Dr. Ehrenstorfer GmbH公司;C18、N-丙基乙二胺(primary secondary amine,PSA)吸附劑 中國Agela Technologies公司。
1.2 儀器與設備
LCQ超高效液相色譜-質譜聯(lián)用儀(配有電噴霧離子源及Xcalibur1.2數(shù)據(jù)處理系統(tǒng)) 美國Finnigan公司;HP 1100高效液相色譜系統(tǒng)(配有可變波長紫外檢測器和Rev.A.06.03色譜工作站) 美國惠普公司;CT18RT型高速臺式離心機 上海天美公司;渦旋混合器 大龍興創(chuàng)實驗儀器有限公司;KQ-500E型超聲波清洗器昆山市超聲儀器有限公司。
1.3 方法
1.3.1 標準溶液的配制
分別準確稱取10 mg羅丹明B、堿性橙II、堿性橙21、堿性橙22、堿性嫩黃標準品,用乙腈定容配制質量濃度為1 mg/mL的單標儲備液,將蘇丹紅I、蘇丹紅II、蘇丹紅III、蘇丹紅IV貯存在棕色儲存瓶中,4 ℃保存?zhèn)溆谩J褂脮r,取各單標儲備液適量,用初始比例流動相(0.1%甲酸溶液-乙腈(95∶5,V/V)配制成不同質量濃度的混合標準溶液。
1.3.2 樣品的前處理
稱取辣椒紅色素樣品2.0 g(精確至0.01 g)于50 mL離心管中,加入10 mL 50%的乙腈溶液渦旋混勻,加入3 g氯化鈉進行鹽析,加入300 mg C18吸附劑凈化后,渦旋混合2 min,超聲提取10 min,以5 000 r/min離心5 min,取上清液。再加入10 mL 50%的乙腈溶液重復提取一次,合并2 次上清液用50%的乙腈溶液定容至20 mL。取1 mL經(jīng)0.22 μm微孔濾膜過濾后進樣測定。
1.3.3 色譜條件
Waters XBridge BEH C18(100 mm×2.1 mm,2.5 μm)分析柱;流動相A為0.1%甲酸溶液,流動相B為乙腈。梯度洗脫程序:0.1~1 min,5% B;1~3.5 min,5%~98% B;3.5~10.0 min,98% B;10.0~10.5 min,98%~5% B。流速0.3 mL/min;進樣量2 μL。
1.3.4 質譜條件
電噴霧離子源;正離子掃描;多以應監(jiān)測(multiple reaction monitoring,MRM);離子源溫度500 ℃;離子源電壓5 000 V;霧化氣、氣簾氣、輔助氣和碰撞氣均為高純氮氣。MRM檢測離子對、去簇電壓及碰撞電壓見表1。

表1 9 種非食用色素的質譜優(yōu)化條件Table 1 Optimized mass spectrometric parameters for 9 inedible pigments
1.4 數(shù)據(jù)統(tǒng)計及圖表繪制
數(shù)據(jù)統(tǒng)計和處理采用液相色譜-質譜聯(lián)用儀的Xcalibur1.2數(shù)據(jù)處理系統(tǒng),圖表繪制采用Microsoft Office Excel 2007版。
2 結果與分析
2.1 提取條件的優(yōu)化
考察常用的提取溶劑乙腈、甲醇、乙酸乙酯、正己烷、乙醚作為提取劑時其提取效果的對比實驗。實驗以樣品中目標物的回收率作為提取效果的評價指標。辣椒紅色素能很好溶于乙腈和甲醇中,但結果表明乙腈提取效果比甲醇好;而正己烷、乙醚和乙酸乙酯作為提取溶劑時,不僅提取液中雜質較多,且提取效果欠佳。所以采用乙腈作為提取溶劑。
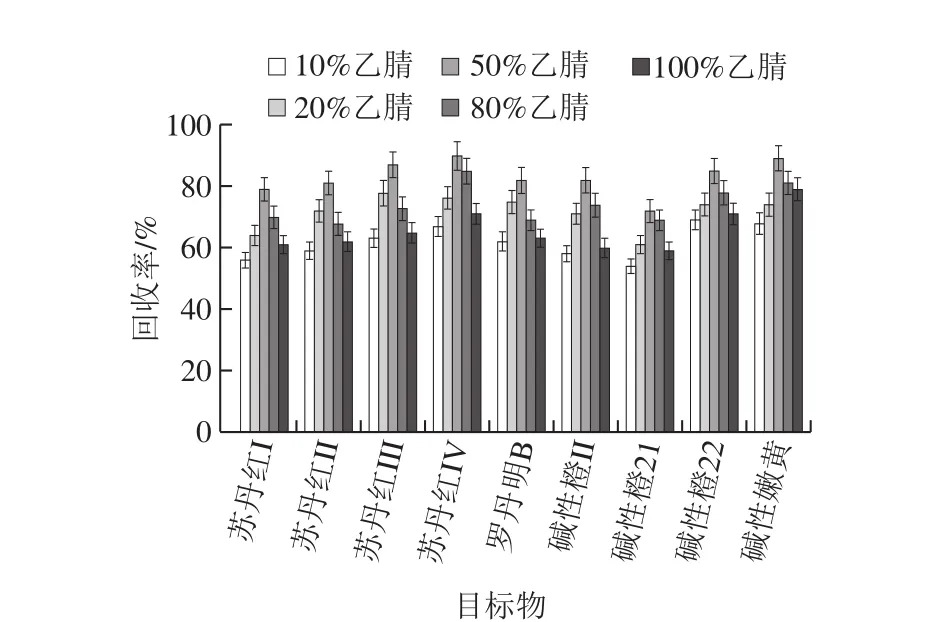
圖1 乙腈溶液體積分數(shù)對目標物回收率的影響Fig. 1 Effect of ratio between acetonitrile and water on recoveries of target compounds
由于辣椒紅色素基質復雜,在乙腈中加入適量的水有利于對辣椒紅色素樣品中雜質的去除,實驗選取體積分數(shù)10%、20%、50%、80%、100%的乙腈溶液作為提取溶劑,結果如圖1所示。50%乙腈溶液提取時具有較好峰形,且9 種非食用色素的回收率均較高,提取效果最好。故選擇50%乙腈溶液作為提取溶劑。
2.2 凈化條件的優(yōu)化

圖2 吸附劑對目標物回收率的影響Fig. 2 Effect of sorbents on recoveries of target compounds
常用的吸附劑有PSA、球形ODS C18填料、NH2氨基填料等[20-28]。辣椒紅色素中富含色素、油脂等成分,根據(jù)辣椒紅色素的基質特點,本實驗選取了PSA、C18以及PSA與C18兩種吸附劑組合作為考察對象用于前處理,如圖2所示。結果發(fā)現(xiàn),PSA對非食用色素會有一定量的吸附作用,C18對辣椒紅色素中油脂等雜質有較好的吸附效果,基質干擾小,且目標化合物的回收率更高。實驗還對C18吸附劑的用量進行優(yōu)化,如圖3所示。結果表明,吸附劑的用量越多吸附效果越好,但過多的吸附劑可以吸附目標化合物使回收率降低,當吸附劑的用量在300 mg時具有較好的吸附效果。
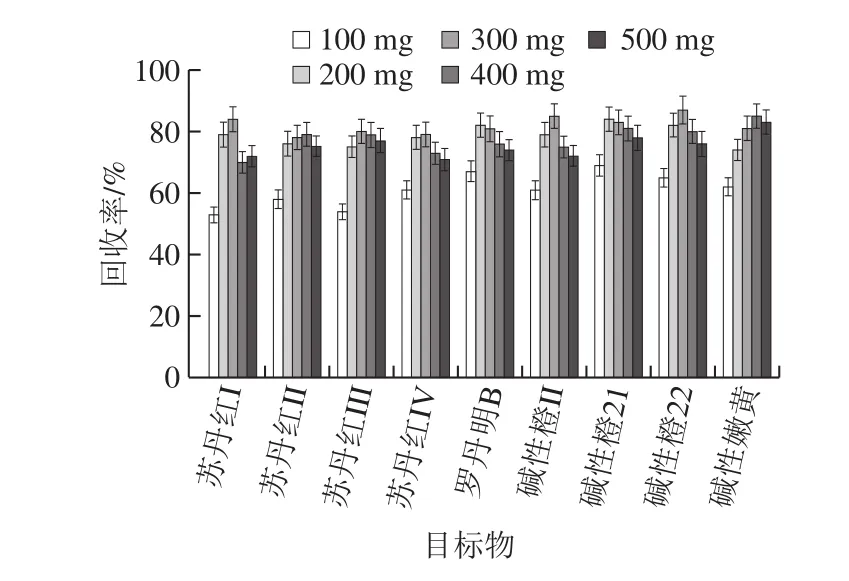
圖3 C18用量對目標物回收率的影響Fig. 3 Effect of C18 dosage on recoveries of target compounds
2.3 超高效液相色譜條件的優(yōu)化
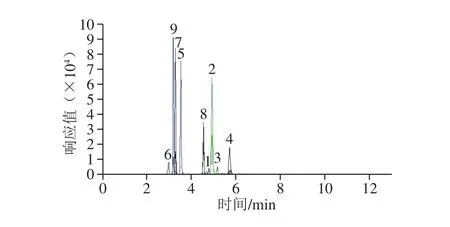
圖4 9 種非食用色素的MRM色譜圖Fig. 4 MRM chromatograms of 9 inedible pigments
9 種非食用色素中的4 種蘇丹紅具有相似的基團和結構,為了準確定量,需色譜峰間有一定分離度。實驗考察甲醇、乙腈、5 mmol/L乙酸銨溶液、0.1%甲酸溶液作為流動相,采用梯度洗脫技術,在C18色譜柱上進行色譜分離,結果表明,甲醇作為流動相時,9 種非食用色素分離度低,峰形較差,質譜響應較低;當以乙腈-乙酸銨和乙腈-0.1%甲酸溶液為流動相時,目標物峰形較好,但乙腈-0.1%甲酸溶液體系靈敏度更高,分離效果更好,如圖4所示。因此,選擇0.1%甲酸溶液-乙腈作為流動相,采用1.3.3節(jié)的梯度洗脫程序。
2.4 質譜條件的確立
非食用色素在質譜的行為較為復雜,將質量濃度為100 ng/mL的蘇丹紅I、蘇丹紅II、蘇丹紅III、蘇丹紅IV、羅丹明B、堿性橙II、堿性橙21、堿性橙22、堿性嫩黃9 種標準溶液,針泵進樣進入質譜系統(tǒng),采用Q1全掃描和碎片離子掃描確定其母離子和子離子,在針泵進樣過程中優(yōu)化碎裂電壓、碰撞電壓等儀器參數(shù)。每種分析物要選擇最強且穩(wěn)定的離子作為定量離子,另一個則為定性離子。9 種非食用色素的質譜條件經(jīng)優(yōu)化后結果見表1。
2.5 方法學驗證
2.5.1 特異性
采用多以應監(jiān)測模式,提取偏差在±0.005‰范圍內(nèi)的精確m/z的色譜圖進行定量,加之色譜保留時間,方法的特異性高,沒有發(fā)現(xiàn)有干擾成分影響9 種目標物的測定。在空白樣品和空白樣品添加的色譜質譜圖中未發(fā)現(xiàn)干擾峰,說明方法具有較好的選擇性。
2.5.2 基質效應、線性關系、檢出限和定量限
1.3.2 節(jié)前處理凈化方法的基質效應進行評價,其結果見表2,其中堿性橙II的基質效應最強,為0.07,其余均在-0.17~0.06之間。雖然9 種非食用色素均有一定的基質效應影響,但并不影響測定的準確性。有文獻研究基質效應在-0.20~0.20之間可以不考慮基質效應的影響[29-31]。
用空白樣品處理液按照1.3.1節(jié)配成系列標準工作溶液,用分析物峰面積對被測組分的質量濃度繪制標準曲線。由表2可知,堿性橙II、堿性橙21、堿性橙22、堿性嫩黃在0.2~8 ng/mL范圍內(nèi)線性關系良好,蘇丹紅I、蘇丹紅II、蘇丹紅III、蘇丹紅IV、羅丹明B在2~40 ng/mL范圍內(nèi)線性關系良好,其線性相關系數(shù)R2高于0.995 0。
以3 倍和10 倍信噪比分別確定了方法的檢出限和定量限,結果見表2。9 種分析物的定量限在0.6~5.0 ng/g之間。

表2 方法的線性、基質效應、檢出限和定量限Table 2 Linearity, matrix effect, LODs and LOQs of the method
2.5.3 準確度和精密度
在3 個水平下做樣品加標回收率實驗,考察方法的準確度和精密度(表3),平均回收率結果在69.6%~92.5%之間,相對標準偏差(relative standard deviation,RSD)在2.8%~8.5%之間。說明方法具有較好的準確度和精密度。

表3 方法的回收率和精密度(n= 5)Table 3 Recoveries and precision of the method (n= 5)
2.6 辣椒紅色素樣品的測定結果

圖5 含有非食用色素的辣椒紅色素樣品Fig. 5 Detection of inedible pigments present in real sample
用實驗建立的方法檢測20 種辣椒紅色素樣品,在其中1 個樣品中含有微量的蘇丹紅染料(圖5),但含量低于我國規(guī)定的最大殘留量1.0 μg/kg。
3 結 論
通過優(yōu)化和對比實驗,探討QuEChERS提取及凈化等前處理條件,考察辣椒紅色素樣品的基質效應,優(yōu)化色譜和質譜條件,研究方法的回收率、檢出限和定量限等技術指標,實現(xiàn)了辣椒紅色素樣品中9 種非食用色素的同時檢測。實驗結果表明,方法的基質效應在-0.17~0.07之間,檢出限在0.2~1.5 ng/g之間,定量限在0.6~5.0 ng/g之間,堿性橙II、堿性橙21、堿性橙22、堿性嫩黃在0.2~8 ng/mL范圍內(nèi)呈線性,蘇丹紅I、蘇丹紅II、蘇丹紅III、蘇丹紅IV、羅丹明B在2~40 ng/mL范圍內(nèi)呈線性,目標物線性相關系數(shù)(R2)均大于0.995 0,3 個樣品加標水平的平均回收率在69.6%~92.5%之間,RSD在2.8%~8.5%之間(n=5)。該方法簡單快捷、準確可靠,適合于市售辣椒紅色素中非食用色素的快速檢測。
