Carvajal綜合征一例伴橋粒斑蛋白基因新突變
2020-06-05 06:53:52吳維鄭璐瑤潘超蘭楊挺李明
中華皮膚科雜志 2020年4期
吳維 鄭璐瑤 潘超蘭 楊挺 李明
1無錫市兒童醫院皮膚科214023;2上海交通大學醫學院附屬新華醫院皮膚科200092
通信作者:李明,Email:liming01@xinhuamed.com.cn
Carvajal綜合征是一種罕見的遺傳性心臟皮膚綜合征,其致病基因為橋粒斑蛋白(desmoplakin,DSP)基因,臨床表現為羊毛狀發、掌跖角化和擴張型心肌病等[1?2],也可伴發白甲病、牙齒異常、聽力減退及反復感染[3?5]。該病的癥狀通常隨著時間的推移而顯現[6]。羊毛狀發可出生既有,掌跖角化一般在嬰兒期顯現,心臟癥狀早期不一定出現[5?6]。我們報道1 例表現為羊毛狀發及掌跖角化但無心臟癥狀的Carvajal綜合征,并對其進行基因檢測。
病歷資料
患兒女,3 歲。出生時頭發卷曲,外觀呈羊毛狀,8 月齡時掌跖部出現點狀角化過度,漸累及膝蓋,無胸痛、心悸、呼吸短促,心電圖、心臟彩超檢查均未見異常。父母體格檢查和心臟彩超檢查均未見異常,非近親結婚,家族中無類似病例。體檢:一般情況可,身高、體重和智力均在正常范圍。頭顱無畸形,心肺肝脾未見異常,四肢肌張力正常,神經系統檢查無特殊。皮膚科檢查(圖1):頭發卷曲,稍稀疏,呈羊毛狀改變,眉毛、睫毛未見異常。掌跖輕度點狀角化過度,膝蓋角化性丘疹,余皮膚未見異常。患兒無牙齒、黏膜、指甲、汗腺等其他外胚層發育異常表現。
基因突變檢測
1. 外周血基因組DNA 提取:經上海交通大學醫學院附屬新華醫院醫學倫理委員會批準(批件號XHEC?D?2019?115),并經患兒父母簽署知情同意書后,分別采集患兒及其父母外周血2 ml。應用QIAamp DNA Blood MinikitⅠ(德國QIAGEN 公司)試劑盒提取全血基因組DNA。同樣方法提取100例無親緣關系的健康個體血樣基因組DNA 作為對照。
2.二代靶向測序和Sanger測序:使用上海安百隆生物科技有限公司設計的遺傳性皮膚病檢測專用Panel(由羅氏公司合成),用IlluminaHiseq X Ten高通量測序儀測序,在相關變異頻率數據庫[寡核苷酸多態性數據庫(dbSNP)、千人基因組數據庫、ClinVar數據庫和人類基因突變數據庫(HGMD)]中對致病變異位點進行評估,根據美國醫學遺傳學與基因組學學會(ACMG)遺傳變異分類指南及患者的臨床表型進行致病變異的篩選。使用Primer?BLAST 工具(https://www.ncbi.nlm.nih.gov/)設計基因 引 物(DSP ? E23:正 向 引 物 序 列 5′ ?AGAGGACTGTGAAGGACCAG?3′和反向引物5′?AAGAACAGCAGGGCACACAG?3′;DSP?E24:正向引物5′?TTTGATGGGCTGAGGAAGAA?3′和反向引物5′?ATTGACAGTGGACGGTCTCA?3′),并通過PCR 擴增,用ABI3730XL 型全自動測序儀進行Sanger測序。
3.測序結果:見圖2。患兒DSP基因第23號外顯子發生移碼突變c.5152dupT(p.L1718Ffs*15),即編碼區第5152 位核苷酸T 重復,導致編碼蛋白第1718 位亮氨酸變為苯丙氨酸,隨后第15 位氨基酸出現1個終止氨基酸;還檢測到第24號外顯子存在無義突變c.C6478T(p.R2160X),即編碼區第6478位核苷酸由C變為T,導致編碼蛋白第2160位精氨酸變成終止氨基酸。患兒母親第23號外顯子亦檢測到移碼突變c.5152dupT(p.L1718Ffs*15),而其第24 號外顯子未檢測到基因突變。患兒父親、100例健康對照中均未檢測到以上2個DSP基因突變。
診斷與治療
根據患兒臨床表現和DSP基因檢測結果等,診斷為Carvajal 綜合征。患兒掌跖角化相對輕微,目前未特殊處理;每半年進行心臟全面檢查(心電圖、動態心電圖監護、心臟超聲等),目前未發現異常。
討 論
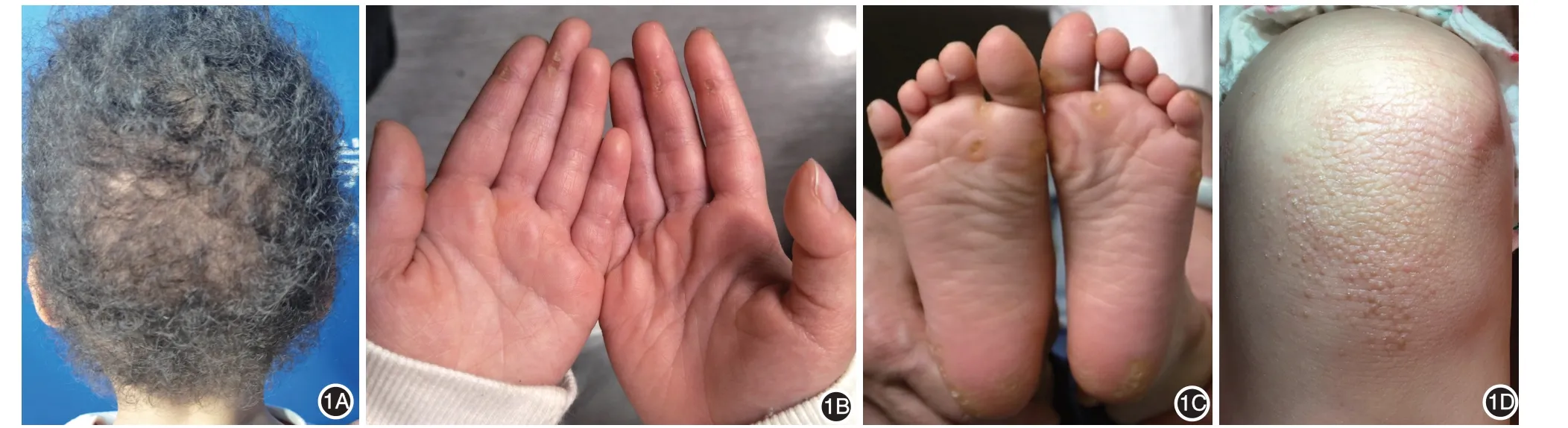
圖1 患兒3歲時,頭發卷曲,稍稀疏,呈羊毛狀改變(1A);掌跖輕度點狀角化過度(1B、1C);膝關節伸側角化性丘疹(1D)
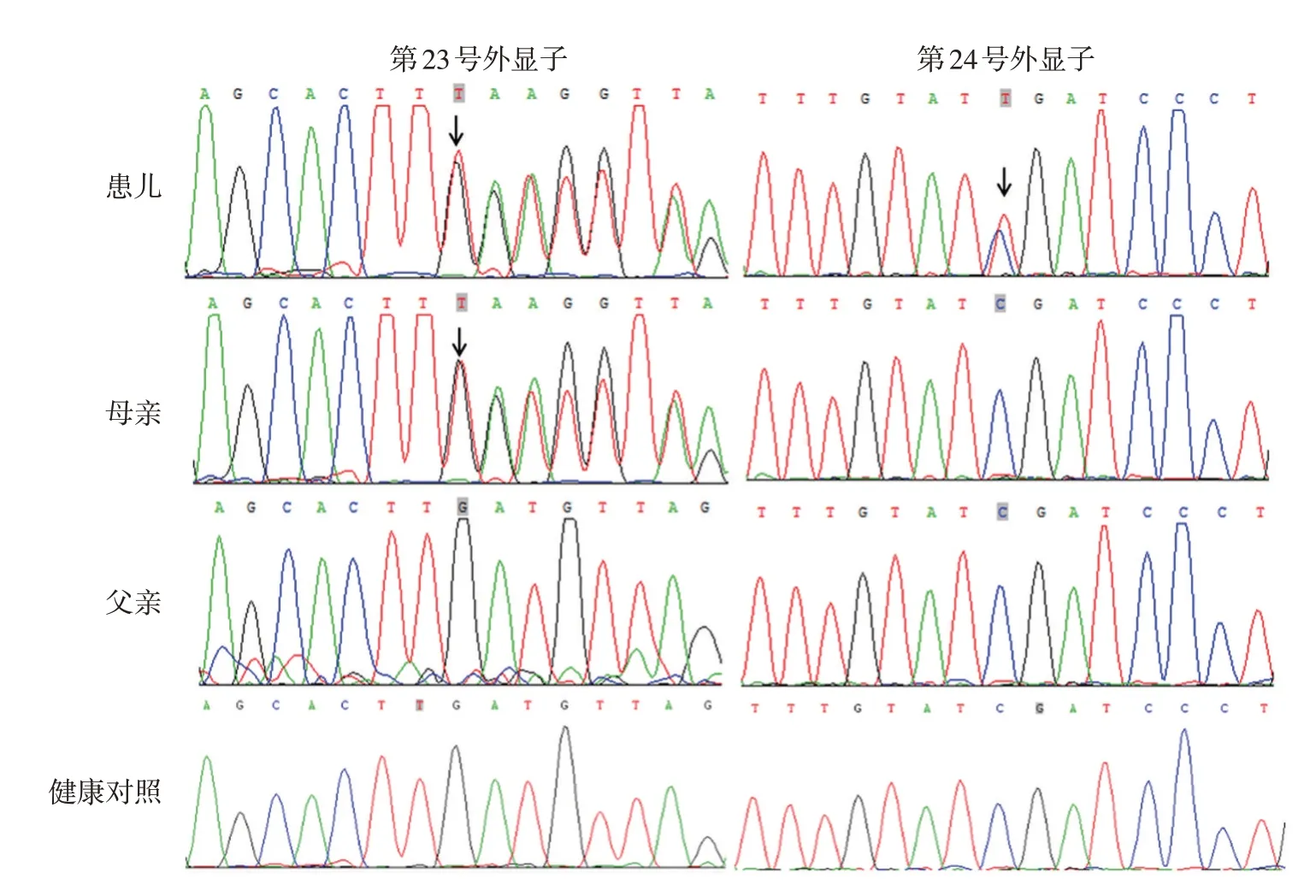
圖2 患兒、其父母及健康對照橋粒斑蛋白(DSP)基因突變分析 患兒DSP基因第23號外顯子發生移碼突變c.5152dupT,第24號外顯子發生無義突變c.C6478T;患兒母親DSP基因第23號外顯子存在移碼突變c.5152dupT,第24 號外顯子未發現突變;患兒父親及健康對照第23、24 號外顯子相應位點均未發現突變
Carvajal綜合征是一種罕見的遺傳性心臟皮膚綜合征,目前國內外報道不多,其中國內中文報道1 例[7]。Carvajal 綜合征可認為是另一種心臟皮膚綜合征Naxos病的一種變異型[8],Naxos病也表現為羊毛狀發、掌跖角化,但心臟表現主要為心律失常性右心室心肌病,致病基因為斑珠蛋白[9]。Carvajal綜合征主要為常染色體隱性遺傳,突變熱點區域主要在DSP基因第23、24號外顯子,少數為常染色體顯性遺傳,主要表型除羊毛狀發、掌跖角化和擴張性心肌病外,可伴有牙齒異常,突變區域主要在DSP 基因第13、14 號外顯子[10]。經二代靶向測序分析,我們發現本例患兒的突變基因為DSP 基因。DSP是橋粒形成過程中的一個錨定蛋白,直接或間接與其他幾個形成橋粒的蛋白相互作用。DSP 基因大致可分為N 端、Rod 區域和C 端3 個主要區域[11],3個區域內都有引起Carvajal 綜合征、皮膚脆性及嚴重棘層松解性大皰性表皮松解癥的突變位點,位于N端的p.Q331*突變可只引起掌跖角化,而C 端p.R2522Sfs*39 和p.R2586*突變的患兒僅毛發和皮膚異常,而無心臟病變[10]。因此,基因型與表型的相關性不易預測[10]。
本例患兒攜帶復合雜合突變,分別為DSP基因第23 號外顯子移碼突變c.5152dupT(p.L1718Ffs*15)和第24號外顯子無義突變c.C6478T(p.R2160X),前者來自于其母親。患兒表型高度符合Carvajal綜合征,且患兒父親及100例健康對照中均未檢測到上述2個DSP基因突變,此2種突變在東亞人群中的突變頻率為0,提示為致病突變,而非單核苷酸多態性。檢索HGMD 數據庫(http://www.hgmd.org)及相關文獻,未見報道移碼突變c.5152dupT(p.L1718Ffs*15)。此突變位于DSP 基因Rod 區域(c.3585?5379或p.1195?1793),該片段僅存在于DSP 異構體Ⅰ(DSPⅠ)中,另外2 種異構體分別為DSPⅡ和DSPⅠa,它們缺少Rod 區中央α-螺旋桿狀結構域的一部分,DSPⅡ缺少氨基酸1195-1793,DSPⅠa缺少氨基酸1351- 1793[10,12]。DSPⅡ是調節角質形成細胞黏附的主要異構體[13],皮膚基底層橋粒蛋白的錯位可間接反映心肌細胞間閏盤的情況[14]。對一DSP 基因突變位點為p.E1493X(位于DSPⅠ)心律失常性心肌病家系研究[15]發現,7例突變攜帶者中僅1例表現出卷發及皮膚異常,且心臟癥狀普遍輕微,皮膚免疫組化染色顯示,DSP 和斑珠蛋白的分布與對照皮膚相當。目前,本例患兒亦未發現心臟異常表現,其母親是無臨床癥狀的c.5152dupT基因突變攜帶者,符合上述文獻的觀點。
本例患兒DSP 基因還存在無義突變c.C6478T(p.R2160X),其父母及100例健康對照者均未檢測到該突變,另外父親的口腔黏膜脫落細胞、毛發、精液中也未發現此突變,提示其為新生突變。此突變位于DSP基因的C末端,是與角蛋白中間絲相互作用的關鍵部位,p.R2522Sfs*39 和p.R2586*突變的患兒僅出現毛發和皮膚異常,無心臟病變[10]。同樣發生在DSP 基因C末端p.G2375A 和p.L2329P 突變的兄妹[6],父母亦無臨床表現,其中先證者男,11歲,表現為局灶性掌跖角化、羊毛狀發和擴張性心肌病,其妹5 歲且有相似的臨床表現,但無心臟累及,其妹4歲后每6個月進行全面的心臟檢查,無任何異常表現。目前,p.R2160X突變在Carvajal 綜合征中未見文獻報道,但在心律失常性右心室發育不良及心律失常性右心室心肌病中為致病性突變[16?17]。目前,仍無法準確預測本例患兒的預后,需要定期隨訪并進行心臟全面檢查,早期發現并進行治療。此外,本例出現的2種突變也存在無義介導mRNA降解的可能[18]。
利益沖突 所有作者均聲明不存在利益沖突
