APP/PS1轉基因小鼠腦內Aβ斑塊沉積增多下調CA3和CA1區域神經元及細胞增殖導致學習和記憶損害
2020-06-08 05:42:10劉婉晴楊婧藝
實用藥物與臨床 2020年5期
劉婉晴,劉 航,楊婧藝,馬 英
0 引言
阿爾茨海默病(Alzheimer′s disease,AD)是最常見的一種癡呆類型,約占癡呆病例的70%[1]。AD典型臨床癥狀是認知功能障礙中所包含的記憶能力進行性下降,神經病理學標志是在腦組織神經元細胞外出現β淀粉樣蛋白(Aβ)沉積形成的神經炎性淀粉斑塊、tau蛋白異常磷酸化所形成的神經原纖維纏結,而Aβ沉積可引起腦實質的損傷,可能與學習、記憶和認知功能障礙過程中神經元丟失有關[2]。神經再生是神經干細胞產生神經元的過程,包括4個階段:神經干細胞的增殖、分化、存活以及成熟[3]。有研究發現,Aβ沉積的AD轉基因小鼠其海馬區神經元的丟失可以引起學習和記憶能力的損害[4]。腦成像顯示,在AD的發展過程中,海馬最易受到影響,萎縮程度最為明顯[5]。因此,AD發生的根本原因在于海馬受到了損害。海馬位于大腦顳葉中的雙側結構,不同區域的功能也不同,其中CA3和CA1區域在學習和記憶中擔任重要作用[6]。有報道,AD海馬CA3和CA1區域神經元丟失導致錐體層萎縮[7],也有研究發現,在AD大鼠海馬CA1和CA3區域顯示出神經元數目增加[8]。本研究主要目的是檢測AD小鼠腦內Aβ斑塊沉積增多對CA3和CA1區域神經元以及細胞增殖的影響,并探討其損傷學習及記憶能力的機制。
1 材料與方法
1.1 動物模型與分組 將雄性12月齡小鼠(均在中國醫科大學動物部購買)分為2組:APP/PS1小鼠5只作為實驗組(AD組),是表達嵌合小鼠/人淀粉樣前體蛋白(Mo/HuApp695swe)和突變型人早老蛋白(PS1)的Tg小鼠,Mo/HuApp695swe可使小鼠產生人類的Aβ沉積;C57BL/6J 野生型小鼠5只作為對照組(C57組)。在實驗前至少10 d將小鼠轉移到動物實驗室以適應環境,在標準條件[12 h光照/黑暗循環,溫度(21±2)℃]下飼養動物,使其自由獲取食物和水。
1.2 水迷宮實驗 檢測小鼠行為學變化,采用Morris水迷宮恒溫游泳池(淮北正華生物儀器設備有限公司)測試小鼠空間記憶能力,試驗在裝有23~26 ℃溫水的圓形水池中進行,水池直徑為160 cm,深70 cm,水深40 cm,分為4個象限,一個9 cm×9 cm圓形平臺置于第4象限水平面下1 cm,攝像機位于水平面上方170 cm處,實驗一共為6 d,前5.5 d為學習訓練,最后1 d下午開始進行測試。迷宮分析軟件記錄小鼠游泳總路程、第4象限活動時間、站臺穿越次數和周邊活動時間等數據,以分析小鼠學習和記憶能力。水迷宮實驗結束后多聚甲醛灌注,收集12月齡小鼠腦組織,隨后石蠟包埋。
1.3 取材 在小鼠進行水迷宮檢測后,麻醉,200 ml 4%多聚甲醛灌注后獲得大腦,放置在70%乙醇內,隨后用二甲苯處理,石蠟包埋,隨后用于免疫組織化學檢測。
1.4 免疫組化檢測海馬及皮質Aβ斑塊沉積 將兩組小鼠石蠟包埋的腦組織置于切片機上,獲得厚度為5 mm的連續冠狀切片,間隔為200 mm,將切下的組織置于載玻片上;進行常規脫蠟,抗原修復,然后磷酸鹽緩沖液(PBS)沖洗3次;為阻斷內源性過氧化物酶活性,將載玻片置于3%雙氧水中,并在室溫下孵育20 min,PBS沖洗;滴加山羊血清(1∶10,Solarbio,#SL038),37 ℃孵育30 min;然后在切片上滴加兔抗β淀粉樣蛋白抗體(1∶400,Cell Signaling Technology,#9888S)放入4 ℃孵育過夜;室溫復溫30 min,PBS沖洗3次;加入二抗工作液:辣根過氧化物酶標記的羊抗兔IgG(1∶200,Solarbio,#SE134),37 ℃孵育30 min,PBS沖洗3次;用二氨基聯苯胺(DAB)顯色試劑盒顯色,蘇木素復染、鹽酸酒精分化、脫水封片。通過連接至光學顯微鏡的相機捕獲圖像,使用Image J軟件計數每只小鼠切片中×20顯微鏡獲取的海馬和皮質中Aβ斑塊沉積的百分比。
1.5 免疫組化檢測CA3以及CA1區域NeuN、ki-67表達 將石蠟包埋的兩組小鼠腦組織置于切片機上,得厚度為5 mm的連續冠狀切片,置于載玻片上。常規脫蠟后進行抗原修復,室溫下用3%雙氧水處理切片以阻斷內源性過氧化物酶,PBS沖洗后孵育一抗:鼠抗NeuN(1∶100,Millipore,America,#MAB377),鼠抗ki-67(1∶200,abbkine,America,#ABM40064);4 ℃孵育過夜,PBS常規沖洗繼而加入二抗:辣根過氧化物酶標記的羊抗小鼠IgG(1∶200,Solarbio,China,#SE131),DAB顯色,蘇木素復染,鹽酸酒精分化,脫水封片。通過顯微鏡相機捕獲圖像,使用Image J軟件計數每只小鼠切片中×40顯微鏡獲取的海馬CA3、CA1區域的NeuN陽性細胞百分比以及ki-67陽性細胞百分比。
2 結果
2.1 兩組小鼠的學習和記憶能力比較 兩組小鼠總路程(圖1A,F=0.788,P<0.05)、第4象限活動時間(圖1B,F=2.333,P<0.05)、站臺穿越次數(圖1C,F=0.025,P<0.05)、周圍活動時間比較差異有統計學意義(圖1D,F=1.507,P=0.05);與C57組小鼠相比,AD組小鼠的行為學測試顯示,學習和記憶能力明顯損害。

圖1 兩組小鼠學習及記憶能力比較
2.2 兩組小鼠海馬及皮質Aβ斑塊密集沉積比較 AD組小鼠Aβ斑塊主要分布在海馬以及皮質區域,呈深棕色,與C57組比較分布更為密集。計數兩組小鼠海馬與皮質Aβ斑塊沉積數量,結果顯示,在海馬區,AD組斑塊沉積數百分比為(5.019%±1.457%),而C57組為(0.176%±0.079%);在皮質區,AD組斑塊沉積數百分比為(7.429%±2.002%),而C57組為(0.571%±0.159%)。兩組比較差異有統計學意義(F=14.073,P<0.05;F=7.761,P<0.05)。見圖2。
2.3 兩組小鼠CA3和CA1區NeuN陽性細胞及ki-67陽性細胞表達比較 NeuN是成熟神經元特異性標記物,檢測NeuN陽性細胞數可以評估CA3、CA1區神經元密度。圖3顯示,與C57組小鼠相比,AD組小鼠CA3和CA1區NeuN陽性細胞數量減少,AD小鼠CA3和CA1區域NeuN陽性細胞百分比分別為(4.769%±1.038%)、(5.029%±1.176%),C57小鼠CA3和CA1區域NeuN陽性細胞百分比為(16.161%±1.516%)、(13.121%±2.356%)(F=2.209,P<0.05;F=4.160,P<0.05),提示AD組小鼠CA3、CA1區神經元丟失較C57組嚴重。
Ki-67是細胞增殖標記物,檢測ki-67陽性細胞數評估CA3、CA1區細胞增殖能力。圖4顯示,與C57組小鼠相比,AD組小鼠CA3和CA1區ki-67陽性細胞數量下降,AD小鼠CA3和CA1區域ki-67陽性細胞百分比分別為(4.872±1.029)%、(4.150±1.239)%,C57小鼠CA3和CA1區域ki-67陽性細胞百分比為(10.551±2.004)%、(8.354±1.458)%(F=3.656,P<0.05;F=0.005,P<0.05),提示AD組小鼠較C57組小鼠CA3、CA1區細胞增殖能力下降。

圖2 兩組小鼠海馬及皮質Aβ斑塊密集分布比較
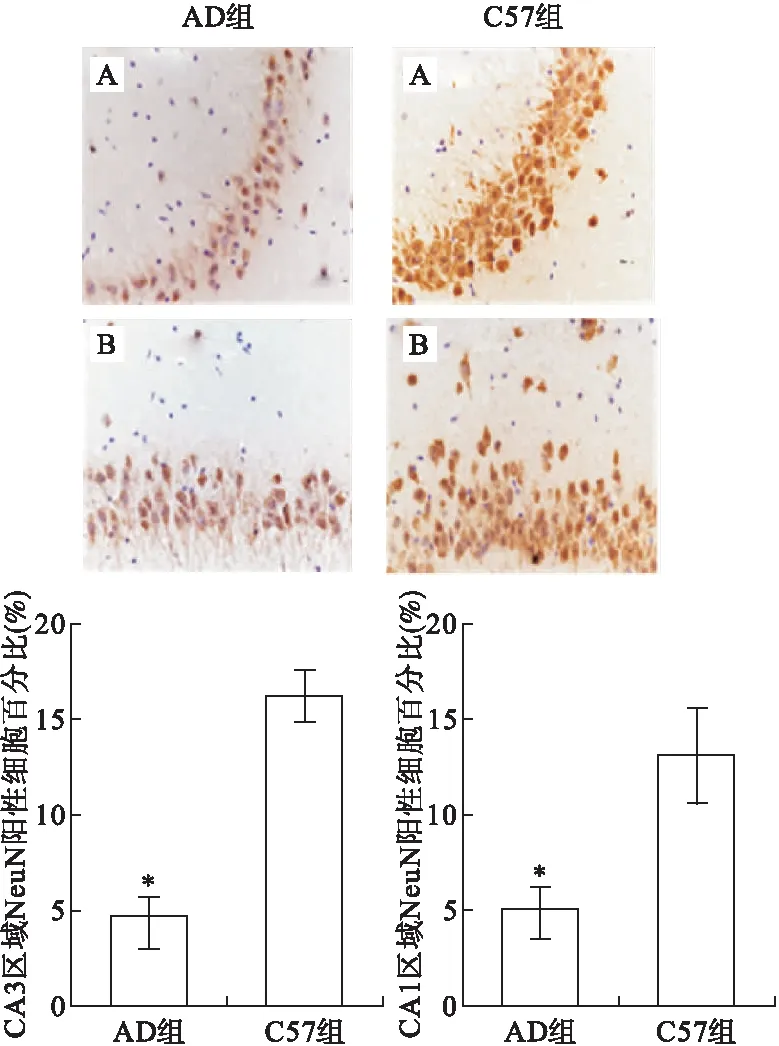
圖3 兩組小鼠CA3和CA1區NeuN陽性細胞表達比較

圖4 兩組小鼠CA3和CA1區ki-67陽性細胞表達比較
3 討論
大腦中的Aβ是由淀粉樣前體蛋白(APP)經β-淀粉酶和γ-分泌酶的連續切割產生。β-分泌酶切割生成APP片段和C99肽,C99肽繼而再由γ-分泌酶切割產生Aβ[9]。Aβ1-42和Aβ1-40是Aβ中最常見的亞型,在腦脊液和血漿中,Aβ1-40是Aβ1-42的10倍和1.5倍。Aβ1-42是AD病理學中的關鍵因素,能夠形成Aβ斑塊沉積的核心。此外,Aβ1-42具有神經毒性,因此,大腦中的Aβ沉積是AD發病機制的早期毒性事件[10-11]。本研究的Aβ斑塊沉積及行為學檢測提示,12月齡AD轉基因小鼠較同月齡C57正常小鼠,Aβ斑塊沉積在海馬及皮質區域密集分布,并且小鼠的空間學習及記憶能力明顯受損。這與Wirths[12]的實驗結果相似,12~14月齡APP轉基因小鼠的神經病理學顯示,海馬中存在大量的淀粉樣沉積物,小鼠學習和記憶能力受損。Unger等[2]的研究結果也發現這一現象,Aβ斑塊沉積在4月齡APP/PS1轉基因小鼠海馬中就可以檢測到,隨著月齡的增長,海馬以及皮質中斑塊負荷顯著增加。我們既往研究發現,Aβ質粒免疫治療可以促進Aβ清除,減輕神經炎癥反應,從而緩解小鼠的空間學習和記憶損害[13],以上研究提示,Aβ斑塊沉積的增多可以引起AD小鼠空間學習和記憶損害。
Aβ沉積可以引起腦實質的損傷,影響最為顯著的是海馬等與智能有關的區域,從而引起學習和記憶障礙[14]。海馬DG、CA3和CA1緊密聚集的神經元構成單向神經回路,研究報道,神經退行性疾病過程中CA3、CA1區域與學習記憶功能息息相關[15]。在海馬注射Aβ1-42可引起CA3區廣泛的神經元變性,表現為CA3中神經元丟失及核收縮,神經元丟失導致AD小鼠腦中學習和記憶能力受損,因此,控制神經元丟失可能是延遲甚至逆轉AD發展的潛在療法[16]。Padurariu等[17]的研究證實,與年齡相匹配的對照組相比,AD患者海馬區神經元密度顯著降低,尤其是在CA3和CA1區域。在APP/PS1轉基因小鼠的研究中發現,早期即出現神經元選擇性丟失,而Aβ是引起神經元丟失的主要因素[18]。本研究表明,AD小鼠Aβ的沉積增多促進小鼠CA3和CA1神經元丟失,進而導致小鼠學習和記憶能力受損。導致神經元丟失的原因存在爭議,體外實驗證明,Aβ促進細胞外Ca2+內流,增加細胞對氧化應激和興奮性毒性的易感性,從而加劇細胞死亡[19]。Aβ斑塊具有毒性,可減少乙酰膽堿(Ach)的合成,合成受阻后導致神經元功能減退或者死亡[20],也有研究報道,在AD小鼠腦內,Aβ的沉積使星型膠質細胞被激活,產生神經炎癥,使神經元數目顯著丟失。
在成年期,神經再生主要發生在以下2個區域:海馬齒狀回的顆粒下層和側腦室的室管膜下區,但是也不排除其他區域依舊存在神經再生情況[21]。增殖的神經干細胞(NSCs)(表達ki-67)分化為中間前體細胞D,D細胞遷移為成神經細胞,經歷4~8周,其生理學和解剖學接近完全成熟的神經元(表達NeuN),接收來自內嗅皮層的信息并投射到CA3區域,進一步傳遞到CA1區域,神經再生過程增加了海馬回路的可塑性以及復雜性[22-23]。Zeng等[24]在APP/PS1/Nestin-GFP三重轉基因小鼠與正常小鼠比較的研究中發現,轉基因小鼠神經干細胞數目減少,未成熟神經元的存活率下降。并且還有研究提示,APP/PS1轉基因小鼠神經再生下調,其神經干細胞增殖下降,神經元減少導致小鼠學習和記憶障礙[25]。但是Jin等[26]發現,AD患者的海馬組織與非AD患者的海馬組織比較,AD患者海馬區成熟神經元的存活數目有所增加,海馬區神經再生上調,上調原因可能是因為機體的一種內源性代償機制,在病理條件下開啟對認知障礙的保護[27],但是隨著時間的推移,這種代償機制是否可以一直持續,尚不明確,有待于進一步研究不同時期的變化。有研究顯示,AD患者海馬的CA1區域ki-67陽性細胞數目增加[28],而本研究中ki-67數目減少,原因可能是檢測ki-67的時間階段不一致。當神經炎癥加劇時,增殖細胞數目可能增加,并且其大部分增殖的細胞可能分化為膠質細胞,隨后神經干細胞增殖能力遠遠低于正常小鼠,ki-67將會迅速下降。本實驗研究檢測CA3、CA1區域ki-67陽性細胞的表達,與C57組正常小鼠相比,AD小鼠ki-67陽性細胞數目下降,提示AD小鼠神經干細胞增殖能力下降,這也可能是相同區域NeuN陽性細胞代表的神經元數目減少的重要原因。神經元丟失是導致AD小鼠腦中學習和記憶受損的不可逆的過程,因此,操控神經再生以補償神經元丟失可能是延遲甚至逆轉AD進展的潛在療法。
綜上所述,與同月齡的C57正常小鼠相比,12月齡AD小鼠腦內有大量的Aβ斑塊沉積,并且海馬亞區CA3、CA1區出現神經元丟失明顯及細胞增殖能力下降,提示下調此區域的神經再生,可導致小鼠學習和記憶能力明顯損害。但是針對Aβ斑塊沉積引發的神經元丟失及神經再生下降的機制尚不明確,是以后研究的方向,調節神經再生將成為緩解甚至逆轉AD的嶄新治療靶點。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
作文周刊·小學二年級版(2022年20期)2022-05-05 01:33:06
學苑創造·A版(2020年9期)2020-10-13 09:41:02
創新作文(小學版)(2019年10期)2019-09-25 08:12:28
作文周刊·小學一年級版(2016年27期)2017-06-03 23:21:17
小學生學習指導(低年級)(2017年5期)2017-05-04 04:14:38
新湘評論·下半月(2016年4期)2016-05-05 22:12:41
新湘評論·下半月(2016年4期)2016-05-05 22:12:41
海外文摘(2016年4期)2016-04-15 22:28:55
作文與考試·小學高年級版(2015年17期)2015-05-30 10:48:04
