線粒體自噬與心力衰竭*
2020-06-09 02:42:18蔣智淵鐘國強
中國病理生理雜志 2020年5期
周 爽, 蔣智淵, 鐘國強
(廣西醫科大學第一附屬醫院心血管內科,廣西南寧530021)
心力衰竭(簡稱心衰)是指由于心臟功能障礙導致心室充盈或射血能力受損的一種復雜的綜合征。目前,我國35歲以上人群慢性心衰的患病率約為0.9%[1]。隨著人口老齡化,冠心病、高血壓、糖尿病等慢性病的發病率增加,以及對急性心血管疾病治療手段的提高,心衰的患病率必然進一步增加。盡管腎素-血管緊張素-醛固酮系統拮抗劑和β受體阻滯劑的應用大大改善了心衰的預后,但心衰患者5年生存率僅為26%~52%[2]。因此,更好地理解心衰的發病機制、尋找潛在的干預靶點,將為慢性心衰提供更多的治療手段。心臟是機體耗能最多的器官之一,線粒體是機體最主要的功能場所,線粒體功能異常與心衰的發生密切相關。而線粒體自噬對于及時清除受損線粒體,維持線粒體正常功能起著重要的作用。為了尋找心衰干預的潛在靶點,本文就線粒體自噬異常在心衰發病中的作用進行闡述。
1 線粒體功能障礙與心衰
線粒體產生心臟最主要的能源物質——三磷酸腺苷(adenosine triphosphate,ATP),這對于維持心臟正常功能至關重要[3]。研究表明,無論是心臟瓣膜病、冠心病、高血壓,還是心肌病所致的心衰患者,均觀察到了線粒體功能異常導致的心肌能量代謝障礙[4]。線粒體功能異常,除了導致心肌能量代謝障礙外,還可以產生大量的活性氧簇(reactive oxygen species,ROS)。ROS可從下述幾個方面促進心肌負性重構,導致心衰:(1)激活蘭尼堿受體2,并抑制肌漿網鈣轉運ATP酶活性,引起心肌細胞鈣超載,肌絲對鈣的敏感性下降,導致心肌收縮功能障礙[5]。(2)促進心肌纖維化:ROS可增加組織金屬蛋白酶抑制劑表達,減少基質金屬蛋白酶表達,促進心肌纖維化;同時,ROS是血管緊張素II介導的心肌肥厚與心肌纖維化的重要下游信號因子[5-6]。(3)激活NOD樣受體蛋白 3(NOD-like receptor protein 3,NLRP3)炎癥小體和Toll樣受體9,增加白細胞介素1β和白細胞介素18等的表達水平,導致心肌炎癥,心肌細胞變性、壞死[7-8]。(4)破壞心肌細胞電穩定性,ROS逆轉Na+-Ca2+交換體功能,導致 Ca2+和 Na+外流;ROS 增加ATP敏感K+內流,縮短心肌細胞動作電位時程;此外,ROS也可以減少KV+內流,增加晚 Na+電流,延長動作電位時程[5]。上述改變破壞心肌細胞電穩定性,與心衰時惡性心律失常及心源性猝死發生密切相關[9]。由此可見,線粒體功能障礙與心衰發生密切相關。
2 線粒體自噬的調控
線粒體自噬是一種選擇性的細胞自噬,是指利用細胞自噬清除細胞內功能異常線粒體的過程。線粒體自噬的調控分為泛素依賴性和非泛素依賴性2種方式。泛素依賴性線粒體自噬包括PTEN誘導激酶1(PTEN-induced kinase 1,PINK1)/E3泛素連接酶parkin介導的線粒體自噬和非parkin依賴性線粒體自噬;而非泛素依賴性線粒體自噬則是指線粒體自噬受體介導的線粒體自噬(圖1)。

Figure 1.Regulation of mitophagy.圖1 線粒體自噬的調控
2.1 泛素依賴性線粒體自噬
2.1.1 PINK1/parkin介導的線粒體自噬 PINK1/parkin介導的線粒體自噬是哺乳動物細胞最常見的線粒體自噬。在正常的線粒體中,PINK1被轉運至線粒體內膜,并被泛素-蛋白酶系統剪切及降解。當線粒體受損傷時,PINK1轉運至線粒體內的過程受阻,在線粒體外膜累積,發生自身磷酸化而被激活。激活的PINK1將parkin招募至線粒體表面并將其磷酸化,磷酸化的parkin將線粒體膜上底物蛋白[如電壓依賴性陰離子通道1(voltage-dependent anion channel-1,VDAC1)、線粒體融合蛋白1(mitofusin 1,MFN-1)、線粒體融合蛋白 2(mitofusin 2,MFN-2)和 mitochondrial Rho(Miro)]多聚泛素化,多聚泛素化的底物被微管相關蛋白1輕鏈3(microtubule-associated protein 1 light chain 3,LC3)適配體蛋白識別,與LC3結合進入自噬體,再與溶酶體結合,形成自噬-溶酶體,最終將受損的線粒體降解[10]。但目前parkin的底物蛋白仍未完全明確。
在介導受損線粒體進入自噬體的過程中,LC3適配體蛋白起著關鍵作用。目前已證實的LC3適配體蛋白有選擇性自噬接頭蛋白p62/sequestosome-1(SQSTM1)、光神經素(optineurin,OPTN)、核點蛋白52(nuclear dot protein 52,NDP52)、TAX1結合蛋白1(TAX1 binding protein 1,TAX1BP1)和BRCA1近鄰基因1(neighbor of BRCA1 gene 1,NBR1)。這些適配體蛋白均有一個LC3結合區域,它們識別線粒體膜上多聚泛素化的底物后,與LC3結合,介導線粒體進入自噬體[10]。
2.1.2 非parkin依賴性的線粒體自噬 除了parkin外,其他E3泛素連接酶,如Gp78、Smad泛素化調節因 子 1(Smad ubiquitination regulatory factor-1,SMURF1)、seven in absentia homolog-1(SIAH1)、線粒體E3泛素蛋白連接酶1(mitochondrial E3 ubiquitin protein ligase 1,MUL1)和 ariadne homologue 1(ARIH1),也可以將線粒體膜上的蛋白底物多聚泛素化,而被p62、OPTN和NDP52識別,從而與LC3結合,進入自噬體。此外,PINK1通過磷酸-泛素化線粒體外膜蛋白,不依賴parkin,直接招募OPTN和NDP52[11]。OPTN和NDP52則進一步招募Unc-51樣激酶1(Unc-51-like kinase 1,ULK1)、含雙FYVE結構域蛋白1(double FYVE domain-containing protein 1,DFCP1)和WD重復結構域磷酸肌醇相互作用蛋白 2(WD repeat domain,phosphoinositide interacting protein 2,WIPI2),介導線粒體進入自噬體[12]。
2.2 非泛素依賴性的線粒體自噬 與依賴線粒體膜泛素化蛋白底物的LC3適配體蛋白不同,有一類線粒體蛋白通過直接與LC3結合,介導受損的線粒體進入自噬體,稱之為線粒體自噬受體。目前發現的自噬受體有Nip3樣蛋白X(Nip3-like protein X,NIX)、BCL2/腺病毒E1B 19 kD相互作用蛋白3(BCL2/adenovirus E1B 19kD-interacting protein 3,BNIP3)、含 FUN14結構域蛋白 1(FUN14 domaincontaining protein 1,FUNDC1)、BCL2樣 蛋白 13(BCL2-like protein 13,BCL2L13)、FK506結合蛋白8(FK506-binding protein 8,FKBP8)、抑制素 2(prohibitin 2,PHB2)和 心 磷 脂(cardiolipin)。 NIX、BNIP3、FUNDC1、BCL2L13和FKBP8位于線粒體外膜,而PHB2和cardiolipin則位于線粒體內膜[10]。
2.3 線粒體動力代謝與線粒體自噬 線粒體通過不斷融合與分裂維持代謝平衡。輕度能量缺乏時,線粒體融合成管狀網絡使產能最大化,滿足機體需要。而當持續、嚴重的應激時,線粒體發生分裂,產生大量的線粒體碎片與ROS,對蛋白、脂質和線粒體DNA造成損害,造成能量供應障礙。線粒體自噬通過清除線粒體碎片,維持細胞穩態。在線粒體動力代謝中,MFN-1和MFN-2介導線粒體外膜的融合,視神經萎縮蛋白1(optic atrophy 1,OPA1)介導線粒體內膜的融合;而發動蛋白相關蛋白1(dynaminrelated protein 1,Drp1)則介導線粒體分裂。研究表明,上述調控線粒體動力學的關鍵蛋白均與線粒體自噬密切相關。MFN-1和MFN-2經parkin泛素化后,可明顯促進PINK1/parkin介導的線粒體自噬。PINK1可間接激活Drp1,促進線粒體分裂,從而更加易于進入自噬體,加速線粒體自噬的進程,抑制Drp1可減弱PINK1介導的線粒體自噬。在FUNDC1和BNIP3介導的線粒體自噬中,OPA1被從線粒體膜中解離,Drp1被招募至線粒體表面并被激活,從而抑制線粒體融合,促進線粒體分裂與自噬。由此可見,線粒體自噬與線粒體動力代謝相互影響,共同維持細胞能量代謝穩態[10]。
3 線粒體自噬與心衰
線粒體自噬能夠及時清除受損的線粒體,避免ROS對細胞的毒性作用,對于維持心肌細胞的穩態起著重要作用。近期,Wang等[13]發現心衰患者心肌細胞線粒體自噬明顯下降。Shirakabe等[14]發現,橫向主動脈縮窄(transverse aortic constriction,TAC)小鼠心衰的發生與線粒體自噬障礙密切相關。Durga等[15]發現,線粒體自噬障礙加速2型糖尿病小鼠心梗后心衰的發生。上述研究提示,線粒體自噬障礙是導致心衰發生的重要原因。線粒體自噬障礙明顯增加ROS水平,損傷線粒體DNA,導致心肌細胞鈣超載、炎癥損傷、壞死和凋亡,以及心肌纖維化,促使心衰的發生[5-8]。但是,心衰時線粒體自噬障礙如何發生?近期的研究證據將為我們提供一些重要的線索。
3.1 線粒體自噬障礙與心衰
3.1.1 PINK1/parkin信號通路與心衰 Wang等[13]發現,心衰患者與TAC心衰小鼠中均存在腺苷酸活化蛋白激酶(AMP-activated protein kinase,AMPK)α2向AMPKα1轉化,同時伴隨線粒體自噬障礙和功能異常。過表達AMPKα2可通過磷酸化PINK1,恢復PINK1/parkin介導的線粒體自噬,從而清除受損線粒體和ROS,減少心肌細胞凋亡,阻止TAC小鼠慢性心衰的發生。Dadson等[16]發現,終末期心衰患者左心室E3泛素連接酶Mule的表達明顯下降。敲除小鼠Mule使得原癌基因c-myc累積,導致PINK1表達下調、線粒體功能異常、ROS生成增多,小鼠出現自發性的心室肥厚、左心室功能障礙。敲除c-myc,可以減少Mule缺失導致的PINK1下調、線粒體功能障礙和心肌損害。Liu等[17]發現,在甲狀腺功能亢進性心臟病中,溶酶體心磷脂酰基轉移酶ALCAT1表達上調,心肌細胞處于氧化應激狀態,出現脂質過氧化、線粒體功能障礙;敲除ALCAT1可上調PINK1,增強線粒體自噬,從而減輕氧化應激、胰島素抵抗,減少甲狀腺素T4造成的心肌損害。此外,Xiong等[18]還發現,過表達PINK1可通過增加線粒體自噬,減少血管緊張素II所致的心肌肥厚效應。
Bhandari等[19]發現,敲除parkin導致果蠅心肌細胞線粒體功能異常、ROS增多和擴張型心肌病的發生。趙佳[20]發現,WDR26過表達可促進parkin向線粒體表面聚集,誘導線粒體自噬,在心肌缺血中起著內源性保護作用。Hoshino等[21]發現,p53與增齡和阿霉素導致的心衰密切相關。p53通過與parkin結合,干擾parkin遷移至受損線粒體的表面,從而抑制線粒體自噬,導致心功能損害。敲除p53,可明顯減少增齡小鼠和阿霉素干預小鼠的心功能損害。Leach等[22]發現,心衰時阻止心肌細胞增生與再生的Hippo信號通路表達上調,通過阻斷Hippo通路中的信號因子Salvador,可以明顯改善心肌梗死小鼠心臟泵功能,而這種作用與增加parkin的表達密切相關。Wang等[23]發現,Hippo通路中的另一信號因子——哺乳動物不育系20樣激酶1(mammalian sterile 20-like kinase 1,Mst1),通過抑制parkin介導的線粒體自噬參與糖尿病心肌病的發生。敲除Mst1增加parkin的表達和心肌細胞清除受損線粒體的能力,減少糖尿病心肌病的發生。
上述研究表明,PINK1/parkin信號通路下調,是導致線粒體自噬障礙和心衰的重要原因,以上調PINK1/parkin信號通路為靶點,改善心肌線粒體自噬有望為心衰的治療提供新的手段。
3.1.2 LC3適配體蛋白/線粒體自噬受體與心衰TAX1BP1是泛素依賴性LC3適配體蛋白,研究表明糖尿病小鼠中,過表達TAX1BP1可通過提高心肌細胞的自噬水平,減少心肌細胞氧化應激、炎癥和凋亡,減輕心肌纖維化和心功能損害,從而減少糖尿病心肌病的發生[24]。
FUNDC1是一種線粒體自噬受體,可直接與LC3結合,啟動線粒體自噬。Zhang等[25]發現,低氧環境可以誘導FUNDC1介導的線粒體自噬,從而抑制血小板活性,減少心肌缺血再灌注損傷;敲除小鼠FUNDC1,導致線粒體自噬障礙,血小板持續性激活,加重心肌缺血再灌注損傷。Wu等[26]發現,心衰患者FUNDC1表達明顯下調;敲除小鼠心肌細胞FUNDC1可導致心肌細胞線粒體擴大和功能異常。Yu等[27]發現,Mst1可通過抑制FUNDC1介導的線粒體自噬,促進心肌細胞缺血再灌注損傷,敲除小鼠Mst1可以明顯減少心肌梗死的擴張區域。另有研究報道,色素上皮衍生因子在缺氧條件下對于心肌細胞的保護作用,與它促進FUNDC1介導的線粒體自噬有關[28]。可見,FUNDC1是重要的心臟保護因子,可通過介導線粒體自噬,維持心肌細胞正常功能,有望成為心衰的干預靶點。
BNIP3也是一種線粒體自噬受體,但與FUNDC1不同的是,目前的研究均提示BNIP3激活與心衰的發生有關。Dhingra等[29]和Du等[30]均發現,BNIP3表達上調與阿霉素誘導的心肌線粒體功能異常及心肌肥厚有關,敲除BNIP3可以減輕心肌線粒體功能障礙和心肌肥厚。Chaanine等[31]發現,BNIP3的表達從大鼠主動脈結扎的第1周開始逐漸增加,發展到心衰階段后到達峰值,并且線粒體數量與BNIP3的表達成反比。阻斷c-Jun氨基末端激酶(c-jun N-terminal kinase,JNK)信號通路,可下調BNIP3表達,減輕心臟壓力負荷增加導致的線粒體丟失和心肌肥厚。BNIP3具有LC3結合區域,當此區域第17和24位絲氨酸殘基磷酸化,促進其與LC3結合,增強線粒體自噬,具有抗凋亡作用。但BNIP3又屬于Bcl-2蛋白家族中的一員,介導細胞和線粒體凋亡,這可能是導致BNIP3激活促進心衰的原因之一[32]。BNIP3的促進線粒體自噬與促凋亡作用如何調控,有待進一步明確,也將是其能否作為心衰干預潛在靶點的關鍵。
3.1.3 線粒體動力代謝、線粒體自噬與心衰 研究表明,線粒體自噬需要Drp1介導的線粒體分裂,分裂的線粒體更加易于進入自噬囊泡,形成自噬體[33]。Shirakabe等[14]發現,抑制Drp1后TAC小鼠線粒體自噬障礙加劇,更早出現心衰。Cahill等[34]也發現,Drp1的功能缺失,導致線粒體自噬障礙,促使心肌炎癥和心衰的發生。可見,Drp1介導的線粒體分裂是維持細胞正常線粒體自噬、預防心衰發的重要調控因素。Bhandari[19]等的發現也驗證上述觀點。他們發現,敲除parkin導致果蠅線粒體自噬障礙和擴張型心肌病的發生,但通過抑制線粒體融合,可以糾正線粒體功能障礙,避免擴張型心肌病的發生,提示在自噬障礙的情況下抑制線粒體融合有可能成為心衰干預的靶點。
但也有研究顯示了相反的結果,Song等[35-36]研究了抑制線粒體分裂對線粒體自噬和心臟的影響。他們發現,敲除小鼠Drp1導致線粒體融合增加,線粒體增大,并出現線粒體過度自噬和線粒體的丟失,最終導致擴張型心肌病的發生。但通過敲除parkin抑制線粒體自噬水平,可以減少心肌壞死、心肌纖維化,糾正心室射血功能。這些研究結果表明,線粒體自噬是一個動態的過程。敲除Drp1的直接作用表現為線粒體自噬抑制,這是早期變化,隨著線粒體不斷融合,體積巨大的異常線粒體必將導致線粒體自噬持續激活、線粒體的過度清除,從而造成心肌能量供應障礙[37]。這也提示,在心衰的不同階段,線粒體自噬所扮演的角色不同,如何精準的調控線粒體自噬是目前亟待解決的問題(圖2)。
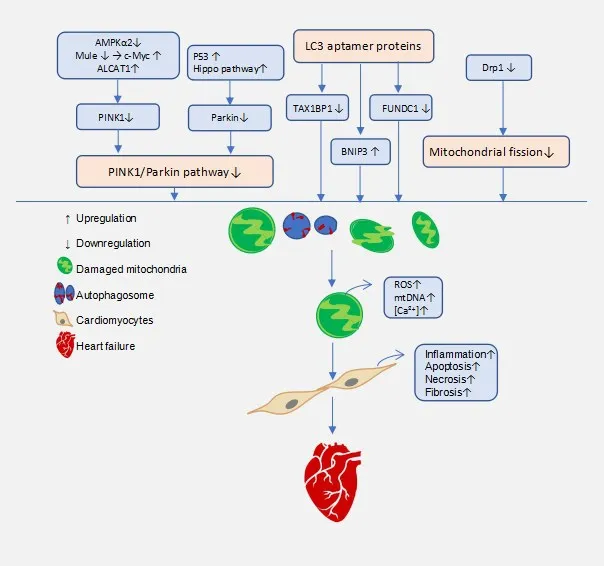
Figure 2.Mitophagy and heart failure.圖2 線粒體自噬與心力衰竭
4 總結
線粒體自噬在心衰發生中作用的闡明,將有利于更好的理解心衰的發病機制,也為心衰治療提供了更多的潛在靶點,具有廣闊的應用前景。但仍有下述問題亟待解決:(1)目前大部分研究均提示線粒體自噬障礙與心衰發生有關,提高線粒體自噬水平可作為治療靶點,但過度自噬也會導致線粒體流失,造成心肌壞死,如何恰當把控線粒體自噬的程度?(2)在疾病的不同階段,線粒體自噬的狀態不同,如何精確調控不同階段的線粒體自噬?(3)目前心衰中線粒體自噬調控的研究,多集中在PINK1/parkin信號通路與個別線粒體自噬受體上,還有較多的LC3適配體蛋白、線粒體自噬受體在心衰中作用有待說明,尤其OPTN和NDP52這兩個最主要的適配體蛋白。(4)線粒體調控是一個十分復雜的網絡,線粒體自噬受體途徑、PINK1/parkin途徑以及線粒體動力代謝之間均存在著交互作用,它們之間的具體關系仍有待進一步研究明確。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
中華詩詞(2022年6期)2022-12-31 06:41:24
學苑創造·A版(2020年9期)2020-10-13 09:41:02
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
中國科技論壇(2017年7期)2017-07-25 08:49:53
媽媽寶寶(2017年2期)2017-02-21 01:21:24
國際漢語學報(2016年1期)2017-01-20 08:21:20
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
中國中醫藥現代遠程教育(2014年22期)2014-03-01 04:32:55
中國中醫藥現代遠程教育(2014年16期)2014-03-01 04:28:54
