UPS-E3泛素連接酶ITCH與胰島素抵抗的關系*
2020-06-09 02:42:18常曉彤
中國病理生理雜志 2020年5期
郭 豪, 馬 玉, 常曉彤
(河北北方學院臨床檢驗診斷學重點實驗室,河北張家口075000)
糖尿病已成為嚴重的世界性公共衛生問題,預計至2045年全球糖尿病患者將超過6億人[1]。胰島素抵抗是2型糖尿病的重要表型,是指各種原因導致胰島素促進葡萄糖攝取和利用率下降,機體代償性地分泌過多胰島素,產生高胰島素血癥。胰島素是一種蛋白類激素,促進糖原、脂質和蛋白質合成等多種生物代謝過程,其作用的發揮依賴于胰島素信號正常轉導;胰島素重要靶器官如肝臟、脂肪、肌肉、胰腺和腦中胰島素信號通路阻斷將導致胰島素抵抗。胰島素信號是一種短暫的效應,信號分子在胰島素的分泌和功能發揮中起著重要的作用。細胞中蛋白質水平的穩態取決于其合成和降解的速率,其中降解過程是通過2個緊密調控的系統來完成的,即溶酶體和蛋白酶體系統,靶向降解錯誤折疊或受損的蛋白質。泛素-蛋白酶體系統(ubiquitin-proteasome system,UPS)是細胞內80%蛋白質降解的主要途徑,其中底物泛素化過程的特異性取決于E3泛素連接酶。已有研究發現E3泛素連接酶ITCH在機體胰島素抵抗中發揮關鍵作用[2]。本文以ITCH的結構和作用機制為基礎,闡述其與胰島素抵抗的相關性,為進一步研究ITCH的功能和胰島素抵抗的機制提供理論基礎。
1 UPS及泛素化
1.1 UPS UPS是真核細胞中選擇性降解蛋白質的主要途徑,在細胞周期、信號轉導、轉錄調控、DNA損傷等多種生物學過程發揮著重要作用[3]。該系統與胰島素信號通路相關,可參與胰島素受體(insulin receptor,IR)的內化、胰島素受體底物1/2(insulin receptor substrate 1/2,IRS1/2)表達量的調控和胰島素的降解[4]。UPS也參與炎癥反應,有報道稱蛋白酶體抑制劑具有抗炎作用,UPS異常與肥胖、胰島素抵抗、糖尿病等多種疾病的發生和發展相關[5-6]。
1.2 泛素化 泛素(ubiquitin,Ub)是一種存在于所有真核細胞、高度保守且大量表達、含76個氨基酸殘基的蛋白分子。泛素化是重要的翻譯后修飾,靶蛋白泛素化首先通過E1泛素活化酶、E2泛素結合酶和E3泛素連接酶的多酶級聯介導、多肽泛素的共價加成而標記,然后經26S蛋白酶體識別并降解,見圖1B。泛素化的調控是由底物特異性的E3泛素連接酶介導[7]。機體通常含有少量E1s和E2s,而含有大量E3s,主要分為HECT、RING和U-box蛋白3種類型。HECT是位于蛋白質羧基端的一個結構域,它的關鍵特征是半胱氨酸殘基在催化底物泛素化前與泛素羧基末端形成中間硫代酯鍵[8]。
2 ITCH及其作用底物
ITCH,又稱atrophin 1相互作用蛋白4(atrophin 1-interacting protein 4,AIP4),是一種高度保守的HECT家族E3泛素連接酶,相對分子質量為113 000,是含有854個氨基酸殘基的單體蛋白,其結構包括:1個N端C2結構域,介導募集的底物蛋白進入細胞膜內;4個WW結構域介導與靶蛋白特異性相互作用,可募集具有絲氨酸/蘇氨酸磷酸化位點或含PPXY結構域的底物蛋白;1個C端HECT結構域可將活化的泛素分子由E2泛素結合酶轉移至靶蛋白;1個PRD結構域位于第195~246個氨基酸殘基之間,參與分揀連接蛋白9(sorting nexin 9,SNX9)等底物蛋白的募集,見圖1A。
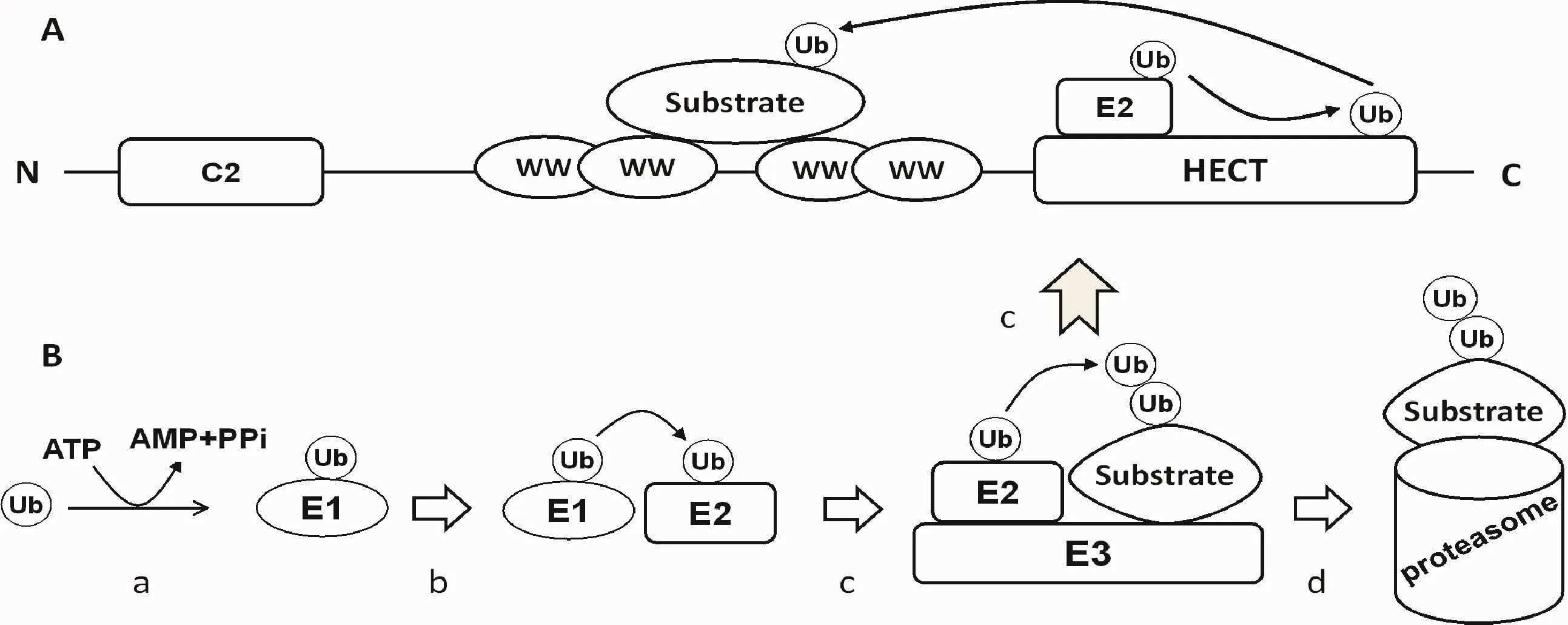
Figure 1.The structure of ITCH(A)and its mechanism of participating in ubiquitin cascade(B).In an ATP-requiring step,a thioester bond is formed between the carboxyl terminus of ubiquitin and an cysteine residue of ubiquitin activating enzyme(E1);activated ubiquitin is then transferred to a specific cysteine residue of ubiquitin conjugating enzyme(E2);the HECT domain of itch binds to the E2-ubiquitin complex and transfers the activated ubiquitin to a catalytic cysteine residue of its C-terminal region,and then catalyzes the ubiquitin transfer from the HECT domain to the lysine residues of the substrates;the ubiquitinated substrates are degraded by the 26Sproteasome.圖1 ITCH的結構及其參與泛素化級聯的作用機制
生理情況下,ITCH的WW結構域與含PPXY基序的底物特異結合,催化其泛素化并經蛋白酶體降解,參與底物介導的免疫調控、細胞周期等多種生物過程。ITCH催化蛋白質泛素化可分為兩步:首先HECT結構域與E2-泛素復合物結合,并將活化的泛素轉移到其羧基端催化半胱氨酸殘基上,然后催化泛素從HECT結構域轉移到底物的賴氨酸殘基上[9]。已有研究顯示,ITCH的作用靶點是多種信號通路的核心參與調控者。
2.1 Jun蛋白家族 c-Jun/JunB是一種活化蛋白1(activator protein 1,AP-1)轉錄因子的成分,現已證實,c-Jun和JunB均是ITCH的底物蛋白[10]。經脂多糖等刺激的巨噬細胞中,炎癥相關基因如白細胞介素 1β(interleukin-1β,IL-1β)的表達依賴于 JunB[11]。在ITCH表達缺失的T細胞中,JunB蛋白表達增加,增強與IL-4結合的活性,后者可通過信號轉導及轉錄激活因子6(signal transducer and activator of transcription 6,STAT6)通路參與抗炎M2型巨噬細胞的代謝及作用的發揮[12]。此外,JunB調節IL-4基因的轉錄,誘導Th2淋巴細胞介導的M2型巨噬細胞極化,產生IL-10等抗炎細胞因子,從而防止肥胖和胰島素抵抗[13]。ITCH靶向結合c-Jun和JunB,催化其泛素化及降解,降低其在防止肥胖相關炎癥和胰島素抵抗中的作用。
2.2 硫氧還相互作用蛋白(thioredoxin-interacting protein,TXNIP) TXNIP最初被認為是一種硫氧還蛋白的抑制劑,現已被報道與胰島素抵抗相關[14]。在動物模型中,TXNIP的高表達可誘導胰腺β細胞凋亡,降低骨骼肌和脂肪等外周組織的胰島素敏感性,通過結合并降低葡萄糖轉運蛋白1(glucose transporter 1,GLUT1)的表達來抑制葡萄糖的攝取[15-16]。機體內質網應激下,TXNIP參與核苷酸結合寡聚化結構域樣受體蛋白3(nucleotide-binding oligomerization domain-like receptor protein 3,NLRP3)炎癥小體的激活,增加IL-1β的生成,進而參與炎癥發生[17]。但是,在細菌感染的巨噬細胞中,沉默TXNIP基因可促進Toll樣受體2介導的誘導型一氧化氮合酶(inducible nitric oxide synthase,iNOS)、腫瘤壞死因子α(tumor necrosis factor-α,TNF-α)和IL-6等炎性因子的產生[18]。ITCH介導的TXNIP在體內外的多泛素化及降解[19],可能靶向TXNIP-炎癥通路,調控機體的炎癥狀態。
2.3 Notch Notch是一種保守的Ⅰ型跨膜受體,它的激活與炎癥、胰島素抵抗等呈正相關[20-21]。實驗表明,肝臟Notch1的表達缺失,誘導糖異生關鍵酶葡萄糖-6-磷酸酶(glucose-6-phosphatase,G6Pase)表達增加,促進葡萄糖產生[22]。另外,Notch激活肝臟中營養敏感的哺乳動物雷帕霉素靶蛋白復合物1(mammalian target of rapamycin complex 1,mTORC1)通路,導致脂肪和甘油三酯生成增多[23]。ITCH通過WW結構域與Notch胞內結構域的N端結合,HECT結構域促進Notch的泛素化[24],從而調控其生物學功能。
2.4 p53蛋白家族 p53是一種轉錄因子,調控細胞增殖與凋亡周期,p63和p73是其同源性轉錄因子[25]。越來越多的證據表明,p53家族成員在調節葡萄糖穩態方面起著重要作用,然而這些作用卻不盡相同[26-27]。TAp63(p63亞型)缺乏的小鼠表現出葡萄糖不耐受、肥胖和胰島素抵抗,而TAp73缺乏的小鼠在高脂飲食喂養下能防止葡萄糖不耐受和胰島素抵抗[28-29]。生理情況下,ITCH催化p63和p73泛素化及降解,使其保持相對較低水平[30],調控p53蛋白家族成員對葡萄糖穩態的影響。
3 ITCH與胰島素抵抗
有學者提出關于胰島素抵抗發生的2種假說,即炎癥假說和信號轉導假說[31]。E3泛素連接酶在胰島素抵抗和糖尿病過程中起著關鍵作用,其機制有兩種:一是E3泛素連接酶通過UPS直接降解IR、IRS和其它關鍵胰島素信號分子;二是通過調節參與胰島素信號分子調節的促炎癥介質間接調控胰島素信號,例如 TNF-α、IL-6、IL-1β、單核細胞趨化蛋白 1(monocyte chemotactic protein 1,MCP-1)、低氧誘導因子 1α(hypoxia-inducible factor-1α,HIF-1α)等[2]。胰島素抵抗的發病機制復雜,是多因素相互作用及影響的結果,包括靶器官細胞胰島素敏感性降低、胞內信號分子轉導受阻、機體代謝紊亂等。
3.1 ITCH靶向炎癥 炎癥聯系肥胖與胰島素抵抗。脂肪組織巨噬細胞和T細胞的活化與肥胖所致的促炎癥狀態有關。M1型巨噬細胞由干擾素γ誘導,分泌促炎性細胞因子,在肥胖脂肪組織中占主導地位,M2型巨噬細胞主要由Th2淋巴細胞分泌的IL-4和IL-13誘導,產生抗炎細胞因子IL-10。已有研究顯示,ITCH-/-小鼠中脂肪組織浸潤的巨噬細胞呈慢性M2型極化,巨噬細胞半乳糖型凝集素2(macrophage galactose-type lectin 2,Mgl2)、精氨酸酶1(arginase 1,Arg1)、幾丁質酶3樣蛋白1(chitinase 3-like protein 1,CHI3L1)等M2型巨噬細胞標志物的表達顯著增加,防止小鼠體重增加及肥胖刺激下的代謝紊亂[13]。體外研究應用siRNA沉默巨噬細胞中ITCH的表達,導致促炎因子IL-1β減少,抗炎因子IL-10表達增加。也有研究表明,ITCH缺乏對肥胖的影響是由高脂飲食引起的炎癥所介導的,當高脂飲食被阻斷時,ITCH基因敲除小鼠在生理過程中體重的變化趨勢與野生型小鼠相同,提示在無高脂飲食或炎癥刺激的情況下,ITCH缺乏不影響小鼠的代謝表型[13]。
炎癥通路通過多種途徑與胰島素抵抗相連,主要有IκB激酶β(inhibitor kappa B kinaseβ,IKKβ)/核因子κB(nuclear factor-κB,NF-κB)和c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)信號通路。ITCH的表達水平與NF-κB轉錄因子的表達水平相關。研究表明,在骨折修復的早期階段,NF-κB結合目的基因啟動子中的相應位點來上調ITCH等含有WW結構域的E3泛素連接酶的表達水平,抑制成骨細胞的分化[32]。含核苷酸結合寡聚化結構域蛋白2(nucleotide-binding oligomerization domain-containing protein 2,NOD2)是一種炎癥性腸病——克羅恩病的易感蛋白,參與協調細胞刺激后的細胞因子應答,其中ITCH直接通過K-63連接多聚泛素化NOD2結合蛋白RIP2,調控NOD2依賴的信號轉導通路,即NF-κB信號通路和Ras-絲裂原激活蛋白激酶(mitogen-activated protein kinase,MAPK)通路,參與NOD2介導的炎癥性疾病的發生[33]。具體地,ITCH增強NOD2/RIP2通路誘導炎癥通路中JNK和p38的活性,但抑制NF-κB的激活。泛素編輯酶A20是炎癥和細胞因子介導的NF-κB激活的關鍵負調節因子,ITCH作為A20泛素編輯復合物的亞基之一,參與A20底物的識別和滅活,負向調控炎癥信號通路[34]。目前已知,JNK與胰島素抵抗的發展相關,機制可能是激活ITCH進一步參與胰島素抵抗。因此,ITCH可通過靶向炎癥通路細胞和分子,調控機體代謝表型和胰島素抵抗的發生。
3.2 ITCH調控機體代謝物質的水平 胰島素抵抗與許多代謝紊亂相關。正常情況下,胰島素通過增加葡萄糖消耗和脂質合成來促進機體合成代謝,但機體發生胰島素抵抗便不能抑制肝臟葡萄糖的產生,自相矛盾的是,肝臟脂質合成增加,導致高血糖和高甘油三酯血癥。
不同胰島素敏感性人群的ITCH表達分析顯示,ITCH與脂肪組織炎癥細胞表型相關,飲食誘導小鼠肥胖的前提下,ITCH基因缺失的脂肪組織細胞體積較小、密度偏高,表現出抗炎模式[13]。經高脂飲食喂養數周后,ITCH-/-小鼠的空腹血糖水平明顯降低,糖耐量得到改善,在空腹和普通飲食狀態下血清胰島素水平下降,胰島素抵抗指數的穩態模型評估值明顯降低[13]。已知IL-13是介導M2型巨噬細胞激活的Th2細胞因子,在ITCH-/-小鼠的肝臟中,抗炎細胞因子IL-13水平升高,激活下游STAT3通路,抑制葡萄糖生成基因的轉錄,從而抑制肝臟葡萄糖的產生[35]。關于脂質代謝,與野生型小鼠相比,ITCH-/-小鼠的肝臟甘油三酯水平明顯降低,過氧化物酶體增殖物激活受體γ(peroxisome proliferator-activated receptorγ,PPARγ)表達顯著上調,后者在組織巨噬細胞中促進M2型極化,增加組織的氧化代謝和能量消耗,改善胰島素抵抗[13]。研究發現,ITCH泛素化并降解脫乙酰酶sirtuin-6(SIRT6),減少脂肪酸氧化并促進肝臟脂質浸潤;另外,ITCH缺失阻止細胞核中固醇調節元件結合蛋白2(sterol regulatory element binding protein 2,SREBP2)的清除,導致循環膽固醇水平降低[36]。將動脈粥樣硬化模型小鼠體內ITCH和載脂蛋白E(apolipoprotein E,ApoE)基因敲除后,M2型巨噬細胞增多,循環斑塊形成減少,同時出現肝脂肪變性減少、線粒體氧化能力增強,脂肪酸氧化增加等脂質代謝改善狀態[36]。總之,ITCH表達缺失背景下,機體甘油三酯、膽固醇等脂質含量減少,血清葡萄糖及胰島素水平降低,胰島素抵抗狀態有所減輕。因此,ITCH的表達調控可以作為治療以高脂血癥和高膽固醇血癥為主要癥狀的胰島素抵抗的一個靶點。3.3 ITCH靶向胰島素及其信號通路 胰島素生物學效應的發揮依賴于細胞中各級信號分子的逐級轉導,其中任何信號分子受抑制或降解,通路被阻斷,即導致胰島素抵抗的發生。既往研究報道,人肝癌HepG2細胞胰島素抵抗狀態下,ITCH蛋白表達上調,可能通過靶向降解胰島素信號通路中關鍵信號分子,抑制胰島素信號轉導[37]。
Gli樣轉錄因子3(Gli-similar 3,Glis3)是Krüppel樣鋅指轉錄因子的一個亞家族,參與調控胰腺β細胞生成和成熟、胰島素基因表達等重要生物過程[38]。ITCH作為Glis3介導的轉錄活性的關鍵負調節因子,可導致其多泛素化并通過蛋白酶體降解而降低Glis3的穩定性,顯著抑制Glis3介導的胰島素基因的轉錄激活,影響胰島素生物學效應[39]。研究表明,ITCH-/-小鼠的肌肉組織中,胰島素受體Tyr972和蛋白激酶B(protein kinase B,PKB/AKT)Ser473的磷酸化程度明顯增強,從而促進胰島素信號轉導[13]。叉頭框蛋白O1(forkhead box protein O1,FOXO1)是胰島素信號通路的重要下游因子,ITCH基因缺失引起底物JunB表達上調以促進miR-182的表達,后者結合FOXO1 3′端非翻譯區,降低了FOXO1的表達[40],抑制其對糖異生關鍵酶的調控,肝臟糖異生作用減低,血糖下降。人表皮生長因子受體3(human epidermal growth factor receptor,HER3)的表達與胰腺癌、乳腺癌等腫瘤的進展相關。抗HER3抗體9F7-F11可誘導ITCH靶向結合HER3,導致HER3泛素化并被蛋白酶體降解。相應地,siRNA介導ITCH基因敲減,可抑制9F7-F11誘導的HER3泛素化降解,恢復HER3磷酸化,促進AKT和細胞外信號調節激酶(extracellular signalregulated kinase,ERK)磷酸化[41],進而促進胰島素關鍵信號通路——PI3K/AKT信號通路的轉導。因此,ITCH介導的胰島素信號分子的降解或信號通路的阻斷可能是胰島素抵抗的分子機制之一。
4 小結
綜上所述,E3泛素連接酶ITCH可直接調控胰島素基因轉錄及其信號通路分子水平,或通過影響炎癥細胞表型和炎癥因子水平間接調控機體甘油三酯、膽固醇等代謝物質水平,進而參與胰島素抵抗的發生。因此,控制機體ITCH蛋白的表達,可能是預防或治療胰島素抵抗和2型糖尿病的新方法。但也有少數報道,如前所述,ITCH作用于底物TXNIP可減輕機體炎癥狀態。目前關于ITCH參與胰島素抵抗的具體分子機制還未明確,有待未來深入研究,為胰島素抵抗相關疾病提供新的理論基礎和實驗依據。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
學苑創造·A版(2020年9期)2020-10-13 09:41:02
中國生殖健康(2019年2期)2019-08-23 08:12:10
電子制作(2018年11期)2018-08-04 03:25:42
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國衛生標準管理(2015年1期)2016-01-14 03:41:27
藥學與臨床研究(2015年4期)2015-06-05 11:35:51
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
