基于多頭注意力機制和殘差神經網絡的肽譜匹配打分算法
2020-06-20 12:01:42王海鵬牟長寧
計算機應用 2020年6期
關鍵詞:模型
閔 鑫,王海鵬,牟長寧
(山東理工大學計算機科學與技術學院,山東淄博 255000)
(?通信作者電子郵箱hpwang@sdut.edu.cn)
0 引言
現如今主流的蛋白質序列鑒定方法中,序列數據庫搜索法[1]因其對不同的質譜類型表現出很強的魯棒性且性能良好而成為最常用的鑒定方法。序列數據庫搜索法的目的是在蛋白質序列數據庫中搜索匹配出最可能產生給定實驗質譜的序列,具體步驟一般分為三部分:對于每一張實驗質譜,首先,從序列數據庫中提取出與產生該實驗質譜的母離子質量偏差在一定范圍內的所有肽段形成肽譜匹配;然后,計算出每一個候選肽段的理論質譜和對應的實驗質譜的得分;最后,再根據分數對肽譜匹配進行排序,取出分數最高者作為鑒定結果。其中,有效準確地計算出肽譜匹配得分對最終的鑒定結果起著決定性的作用。大多數打分算法都依賴于將候選肽轉換成理論質譜,然后計算理論質譜與實驗質譜的某種相似度分數,再通過全局信息進行二次打分。目前比較新的常用數據庫搜索鑒定工具主要包括Comet[2-3]、MSGF+[4]、Mascot[5]、MaxQuant[6]等,其中Comet 和MSGF+作為代表性的開源鑒定工具被廣泛使用,但是這些傳統的鑒定工具中所使用的肽譜匹配打分方法都依賴于已知的肽碎裂規律及相關信息,存在著理論質譜準確度和相似度計算方法有效性等限制。
使用非深度學方法解決肽譜匹配打分問題一直是一個備受關注的問題,Bai 等[7]使用最大二分匹配模型進行肽譜匹配打分并取得了不錯的效果,但由于最大二分匹配模型存在的問題導致肽譜匹配打分仍舊存在很大的問題。為改進此類問題,Bai 等[8]又提出了子模塊廣義匹配(Submodular Generalized Matching,SGM)模型,該模型在匹配準確率上有一定的提升,但是其充分利用質譜離子峰所攜帶的信息以及肽碎裂規律的能力還略顯不足。深度學習領域不斷的發展與完善,也為解決蛋白組學中的一些問題提供了新的途徑。許多深度學習模型被用來解決蛋白組學的一些重要問題,如利用深度學習預測蛋白質結構[9-10]。蛋白組學中的許多問題可以看作是序列信息處理的延伸和拓展,而序列信息處理在深度學習領域中也是一個備受關注的問題。循環神經網絡常常被用作序列信息處理,Zhou 等[11]提出了基于雙向長短期記憶網絡(Bidirectional Long Short-Term Memory network,Bi-LSTM)的碎片離子預測模型用于預測理論質譜,該模型具有良好的預測性能,但是并未進行進一步的肽譜匹配打分實驗。隨著深度學習的發展,眾多學者發現ResNet(Residual neural Network)[12]不僅能有效解決計算機視覺領域許多重要問題,也能被用來有效地處理序列信息,并且已經有一些學者成功將ResNet應用到了蛋白組學的相關領域[13-14]。在現今的自然語言處理領域,許多取得不錯效果的深度學習模型都使用注意力機制[15],注意力機制的核心目標是從眾多信息中選擇出對當前任務目標更關鍵的信息加以利用。在肽碎裂事件中,某一肽鍵的斷裂不只與相鄰兩個氨基酸相關,也會與其他位置的氨基酸存在一定的關聯,因此利用注意力機制能有效地利用這樣一些相關性,進而提高模型的準確度。而多頭自注意力機制(multi-head self-attention mechanism)[16]是對普通注意力機制的改進。因此本文提出了基于深度學習的打分算法:deepScore-α。該算法使用ResNet 與多頭注意力機制相結合的模型用于肽譜匹配打分,有效地結合了兩者優點,利用深度學習自動學習肽碎裂相關規律,能獲得比較好的打分效果。
1 模型與算法
1.1 deepScore-α算法打分流程及模型結構
deepScore-α 打分算法使用深度學習模型學習肽碎裂規律對肽譜匹配進行打分,輸入為肽譜匹配(Peptide Spectrum Match,PSM)中肽序列以及該肽序列在對應譜圖中已標注的碎片離子離散化相對強度。算法分為兩個階段,輸入的肽序列通過特征提取算法轉換為特征序列與肽序列對應的碎片離子離散化相對強度一同輸入模型,不同階段輸入相同,但是會產生不同的輸出(具體流程如圖1所示)。
1)在訓練階段,模型輸出預測的碎裂離子離散化相對強度以評估模型學習肽碎裂規律的效果,并使用交叉熵作為模型損失進行權重更新。
2)在打分階段,模型輸出肽序列對應的碎片離子相對強度概率分布矩陣,再結合實際的碎片離子相對強度計算最終的肽譜匹配分數,而不是給出預測的碎片離子相對強度。
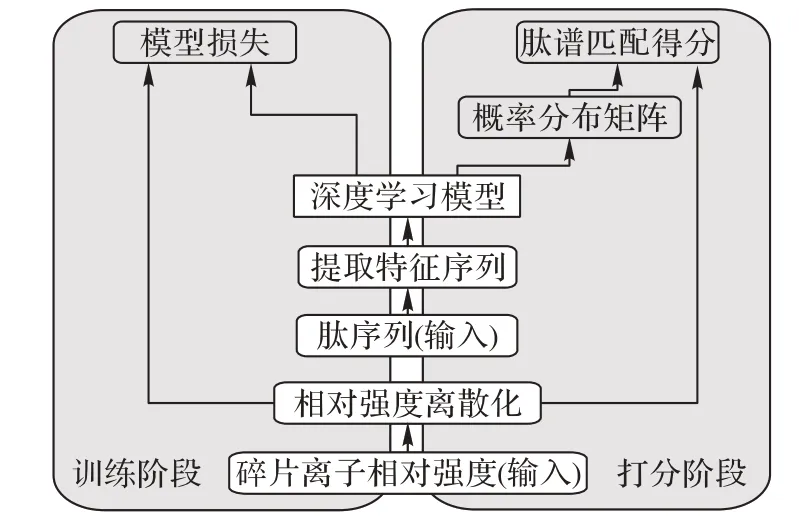
圖1 deepScore-α打分算法流程Fig.1 Flowchart of deepScore-α scoring algorithm
deepScore-α 使用的模型由一維ResNet 模型和多頭自注意力層組成:ResNet由卷積層和殘差鏈接構成,主要用于提取和處理序列特征,多頭自注意力機制則利用注意力機制的特性更加全面準備地學習肽碎裂規律以提升模型預測效果。ResNet 是深度卷積網絡的改進,深度卷積網絡隨著網絡層數的不斷增加,在網絡能夠收斂的前提下,由于網絡優化問題其表現會出現下降。ResNet通過殘差連接能將較淺的網絡層的輸出直接輸入到較深的網絡層,在一定程度上解決了這個問題。deepScore-α 中所使用的ResNet20 參考了He 等[12]提出的ResNet,并且在此基礎上進行了一些改進以適應肽序列信息處理,將其改為一維卷積且控制輸入輸出長度保持不變。
多頭自注意力機制由谷歌于2017 年提出,不同于之前的注意力機制,多頭自注意力機制采用了更加新穎的multi-head機制,即多次計算的注意力結果進行拼接,再通過線性變換獲得最終的多頭注意力結果。在長距離依賴上,因為自注意力需要計算每個輸入位點與其他所有位點之間的關聯,所以能夠忽略位點之間的距離影響從而完整地學習到一個序列的內部結構信息。本文對多頭注意力層中頭的數量對最終模型預測結果的影響進行了比較實驗,選取能產生最優實驗結果的結構作為deepScore-α 最終的模型結構。本文所使用的multihead self-Attention ResNet20 如圖2 所示,最終輸出的b、y 代表b離子和y離子的離散化相對強度。
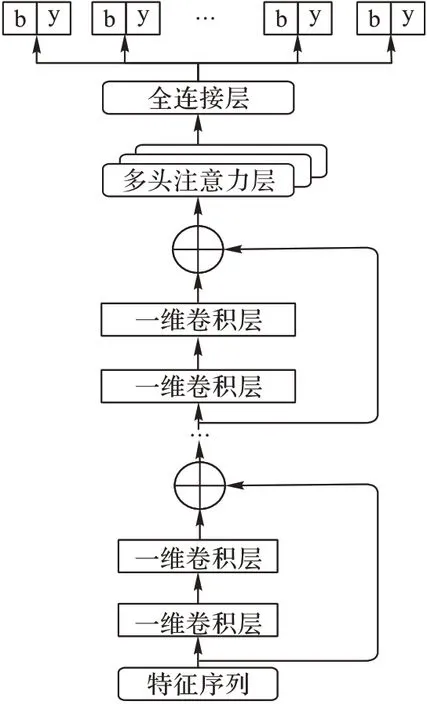
圖2 deepScore-α模型結構Fig.2 Structure of deepScore-α model
1.2 deepScore-α分數計算方法
deepScore-α 所使用的打分算法是一種基于概率和的打分方法。假設候選肽氨基酸序列為X={A1,A2,…,An}(其中n為肽序列總長度),通過特征提取后獲得對應的特征向量F={x1,x2,…,xn-1}。將特征向量序列作為輸入,通過模型獲得概率矩陣P。使用候選肽序列在譜圖中進行譜峰標注,獲得b離子及y 離子的相對譜峰強度N={n1,n2,…,no}(o為該肽序列產生的碎片離子總數),再通過以下公式進行離散化:

得到最終b/y 離子對應的離散化相對強度序列Y={y1,y2,…,yo}。使用以下公式計算出最終的肽譜匹配得分:
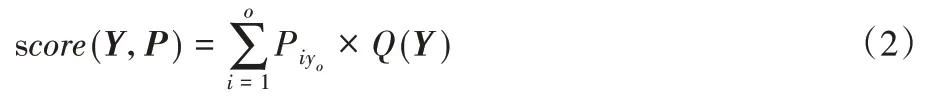
其中:n為肽序列產生的所有b/y離子總數;P為模型輸出的碎片離子相對強度分布概率矩陣,Piyo表示第i個碎片離子實際的離散化相對強度yo在概率矩陣中對應的概率;Q(Y)為肽譜匹配質量系數計算函數。Q(Y)具體計算式如下:
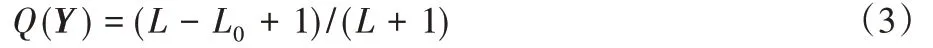
其中:L為離散化相對強度序列Y的長度,L0為其序列中相對強度為0的數量。
1.3 Comet及MSGF+打分算法
本文使用開源鑒定工具Comet以及MSGF+作為對比。其中,Comet 工具所采用的打分算法將實驗質譜譜圖轉換為稀疏矩陣,并使用了快速交叉關聯(Fast Cross Correlation)算法來計算實驗質譜和理論質譜的相似度得分,再通過得到的相似度得分結合全局信息計算出e-value 作為最終肽譜匹配得分;MSGF+工具所采用的打分算法則將肽序列轉化為肽序列向量,再使用概率模型將質譜轉換為質譜向量,通過計算肽序列向量與質譜向量的點乘給出最終的肽譜匹配得分。以上兩種鑒定工具所使用的打分算法均可看作是一種計算肽序列與實驗質譜相似度的打分算法,本文提出的deepScore-α 則通過深度學習模型獲得肽序列對應的碎片離子相對強度的概率和進行打分,無需計算肽序列與實驗質譜的相似度,打破了相似度計算方法的限制。
2 實驗與結果分析
2.1 數據集介紹及處理
本文用于模型訓練的數據集為人類蛋白組數據集(HumanProteome),來自Wilhelm 等[17]在2014年關于人體蛋白組鑒定的相關工作(PRIDE 數據集標識符:PXD000865),包含人體26 個組織的蛋白質二級串聯質譜譜圖及相應的鑒定結果。在一般的肽序列鑒定流程中,蛋白質樣品首先通過質譜儀獲得原始質譜譜圖數據(數據文件格式通常為raw),再使用相應的鑒定工具給出譜圖的對應得分最高的肽段作為鑒定結果。由于鑒定軟件給出的鑒定結果仍會存在錯誤,于是本文通過設置閾值條件q-value≤0.001 及PIF≥0.7 對鑒定結果進行過濾,并且在譜圖存在多個鑒定結果時選取后驗錯誤率(Posterior Error Probability,PEP)較低者作為最終的鑒定結果以保證鑒定結果的可信度(q-value、蛋白質水解誘導因子(Proteolysis Inducing Factor,PIF)和PEP 均為相應的統計學指標)。碰撞能量(Norm Collision Energy,NCE)為質譜儀器一種重要參數,本文使用碰撞能量對該數據集進行劃分,為確保模型最終的訓練效率,去除掉數據量過少的碰撞能量為40 的部分,最終獲得兩個訓練數據集:790 271 條NCE 為30 的肽序列數據集,101 847 條NCE 為35 的肽序列數據集。隨后使用標注誤差限(碎片離子理論質荷比與實際譜圖種譜峰質荷比的差值)為20 ppm 的條件對過濾處理后得到的數據集進行標注,獲得肽序列對應的碎片離子譜峰強度,肽段碎裂會產生多種碎片離子,本文只考慮占比較大的b/y型碎片離子。
在進行打分效果評估時,對所有的原始譜圖文件(raw)進行抽樣,并利用pParse(v2015,一種譜圖提取工具)提取出抽取得原始譜圖文件中的譜圖,再使用Comet(v2018014)和MSGF+(v2019.07.03)對提取的譜圖進行鑒定。由于本文只針對肽譜匹配打分算法進行研究,所以為保證算法效果評估的有效性,在進行打分時對鑒定工具輸出的每張譜圖的前50個較高分數候選肽段進行重新打分。在進行打分Top1 命中率(在已知正確肽序列的情況下正確肽序列在該譜圖所有候選肽中得分最高的譜圖所占比例)實驗時,為了保證命中率計算結果的準確性,分別從碰撞能量為30和35的模型測試集中抽取10 000張原始鑒定結果可信度比較高的譜圖進行打分實驗。用于算法泛化性能測試的數據集來自ProteomeTools2(PRIDE 數據集標識符:PXD010595)[18],同樣對所有原始譜圖數據進行隨機抽樣,再使用Comet和MSGF+進行鑒定,最后使用deepScore-α 對Comet 以及MSGF+輸出的候選肽段進行重新打分并評估打分結果,由于ProteomeTools2數據集原始鑒定結果較為準確,所以直接進行Top1命中率評估。
2.2 特征提取及模型訓練結果
在自然語言處理(Natural Language Processing,NLP)中,對單個字或詞進行編碼是十分重要的,因此逐漸發展出了WordEmbedding[19-20]這樣的特征編碼方式。而本文所涉及的肽碎裂事件中肽鍵可以被看作一個基本單位,即可以借鑒自然語言處理中的編碼方式對其進行特征編碼,通過類似onehot 類型的編碼將氨基酸殘基表示成22 維的向量。在肽鍵碎裂過程中,肽鍵左右相鄰的氨基酸種類及肽序列中堿性氨基酸和肽序列所帶電荷的數量對肽鍵的碎裂起著至關重要的作用。本文綜合考慮了以上因素,最終形成了以下105 維的特征集(以某一肽鍵為例),具體特征集如表1所示。

表1 肽鍵特征集Tab.1 Feature set of peptide bonds
本文也探索了多頭自注意力機制中頭(head)的數量對最終模型的準確率產生的影響,在人類蛋白組數據集上分別進行了不同head 數量的實驗,并且與邏輯回歸以及支持向量機(Support Vector Machine,SVM)進行了比較,實驗結果如表2所示。實驗結果表明,本文采用的模型預測準確率明顯優于邏輯回歸和支持向量機,更有效地學習到了肽碎裂規律,且在不加入多頭注意力機制時ACC(ACCuracy,預測的相對強度值與真實相對強度值相等的樣本占所有樣本的比例)為81.56%;加入多頭注意力機制后,隨著head數量的增加,模型預測結果逐步提升;當head 為8 時,模型準確率達到最優,相較于未加入注意力機制的測試結果提升了約2.6 個百分點;隨著head 數量的進一步增多,模型準確率出現下降,于是deepScore-α后續采用了head為8的模型進行打分實驗。

表2 不同模型在測試集上的預測準確率Tab.2 Prediction accuracy of different models on test set
模型訓練中學習率設置為0.000 1,使用Adam 優化器,為避免模型過擬合,權重衰減設置為0.000 1,最終在測試集上測試結果如圖3 及圖4 所示。因為模型在訓練時預測的是離散化的碎片離子相對強度,所以預測值與真實值相差大小為±1 時也可以認為預測有效。因此在評估模型預測效果時,除了將傳統的ACC 作為評價標準,還需要考慮到預測值與真實值偏差范圍為±1 的部分,從測試結果可以看出,模型在兩個數據集上均獲得了不錯的預測效果,ACC 與±1 的部分所占比例之和都達到了97%。
2.3 肽譜匹配質量系數
對于某一實驗質譜譜圖,產生該譜圖的正確肽序列相較于其他錯誤的候選肽序列往往能在譜圖中標注出更多的非零相對強度的離子峰,有更多的相對強度不為零的碎片離子存在,即非零相對強度的占該肽序列產生的所有碎片離子數量的比例更大。由于deepScore-α 利用碎片離子離散化相對強度的概率和對候選肽進行打分,因此當正確肽序列長度短于錯誤候選肽序列時,即使錯誤候選肽序列有更多的相對強度為零的碎片離子,其概率和也可能超過正確肽序列。本文提出了肽譜匹配質量系數(PSM Quality Coefficient,PQC)用于降低肽序列長度及譜峰相對強度為零的碎片離子對打分算法的影響。
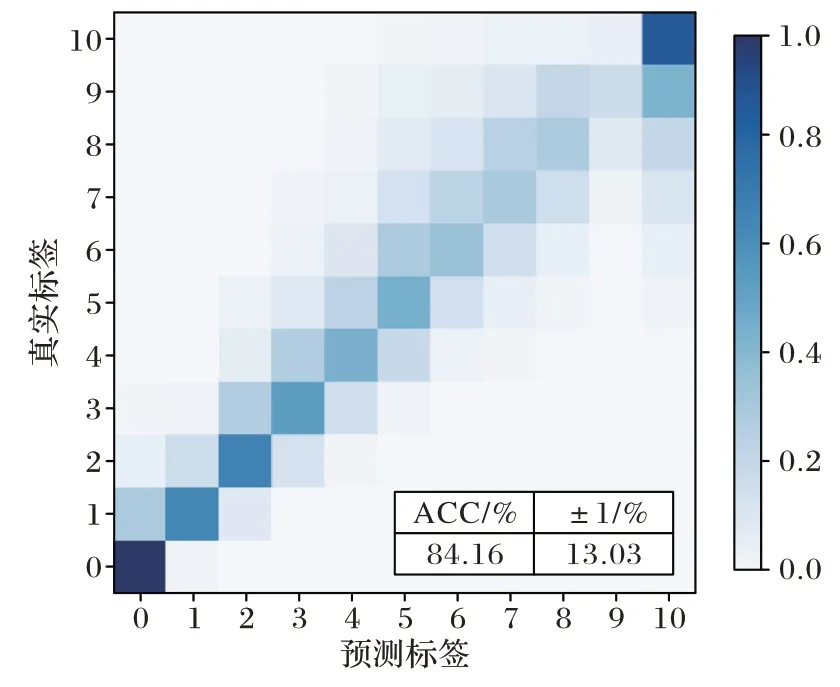
圖3 碰撞能量為30的數據集上模型訓練結果Fig.3 Model training results on dataset with NCE of 30
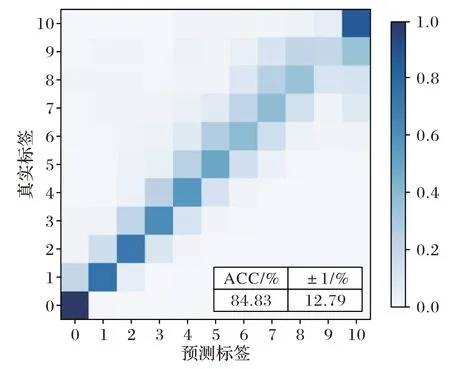
圖4 碰撞能量為35的數據集上模型訓練結果Fig.4 Model training results on dataset with NCE of 35
2.4 deepScore-α與Comet以及MSGF+的打分效果比較
本文使用Comet和MSGF+對碰撞能量為30及35的數據集中的隨機抽取的原始數據文件(raw)進行鑒定,再使用deepScore-α對Comet以及MSGF+輸出的前50條鑒定結果進行重新打分并排序,最終的錯誤發現率(False Discovery Rate,FDR)曲線如圖5和圖6所示。通過比較可以看出,deepScore-α在FDR=0.01時保留的肽序列數量相較于Comet和MSGF+最高提升了約14%,在30碰撞能量的情況下完全優于Comet和MSGF+,而在35碰撞能量的情況下某些部分基本與Comet和MSGF+持平。其原因應該是35碰撞能量的訓練數據過少導致,30碰撞能量最終用于訓練測試的包含790271條肽序列,而35碰撞能量訓練集只有101847條肽序列,并且由于deepScore-α是利用概率進行打分,其對標簽分類的概率更加敏感,因此交叉熵(Cross Entropy)更能反映模型對肽碎裂規律的學習程度。在最終用于打分的模型中,碰撞能量30的模型在測試集上的最小交叉熵為35.92,而在碰撞能量35中測試集的最小交叉熵為40.84,所以在利用概率進行打分時,deepScore-α在35碰撞能量的數據集上的表現比30碰撞能量差一些,如果能有效地擴大35碰撞能量的數據集規模,使其與30碰撞能量的數據集規模一致,使交叉熵降低至相同水平,其最終打分表現也應該一致。
在打分階段比較打分算法有效分辨正確候選肽與錯誤候選肽的能力時,評估比較其Top1 命中率是十分有必要的。本文從劃分為模型測試的數據集中隨機抽取10 000 張譜,再使用Comet 和MSGF+對這10 000 張譜圖進行重新打分,deepScore-α 對Comet 和MSGF+輸出前50 的鑒定結果進行重新打分排序,最終的Top1 命中率比較如圖7 所示。經過比較發現,deepScore-α 的Top1 命中率在30 碰撞能量以及35 碰撞能量的情況下都比Comet 和MSGF+更高,在30 碰撞能量的情況下更高出了5 個百分點,因此deepScore-α 識別正確肽序列的能力要強于Comet和MSGF+。

圖5 deepScore-α、Comet和MSGF+在碰撞能量為30的人類蛋白組數據集上的FDR曲線Fig.5 FDR curves of deepScore-α,Comet and MSGF+on humanbody preteome dataset with NCE of 30
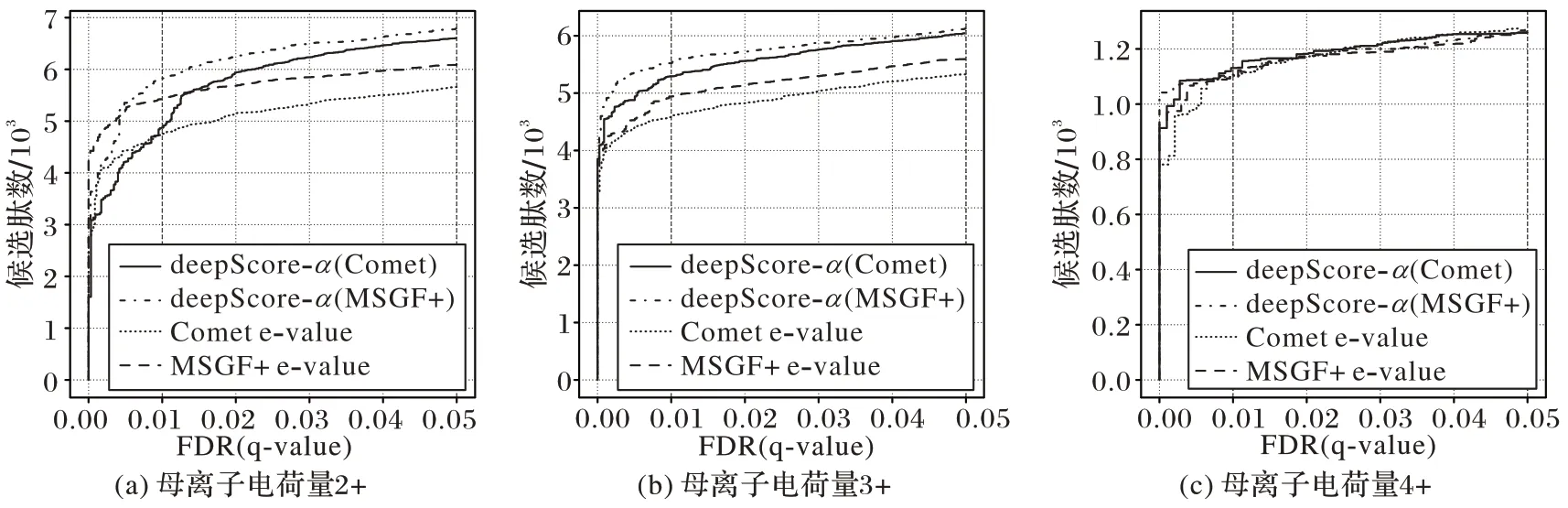
圖6 deepScore-α、Comet和MSGF+在碰撞能量為35的人類蛋白組數據集上的FDR曲線Fig.6 FDR curves of deepScore-α,Comet and MSGF+on humanbody preteome dataset with NCE of 35

圖7 deepScore-α、Comet和MSGF+打分結果中正確候選肽的分布Fig.7 Distribution of correct candidate peptides in deepScore-α,Comet and MSGF+scoring results
2.5 deepScore-α泛化性能分析
打分算法的泛化性能是十分重要的,好的泛化性能能極大地提高打分算法在不同數據集中的表現而不只是在特定某一數據集下表現良好。本文首先在Humanbody數據集上訓練模型,再使用訓練完成的模型對ProteomeTools2 中隨機抽取的譜圖進行打分并與Comet和MSGF+進行比較以評估其泛化性能。同樣先使用Comet 和MSGF+進行鑒定,再使用deepScore-α 對二者輸出的前50 條鑒定結果進行重新打分和排序,FDR 曲線如圖8 和圖9 所示。通過分析比較發現,deepScore-α 在FDR=0.01 條件下保留的肽序列相較于Comet和MSGF+最高提升了約7%,在30 碰撞能量下完全優于Comet,在35 碰撞能量下存在部分與Comet 基本持平的情況,與在Humanbody 數據集中進行打分的表現一致,應該為35 碰撞能量數據量相較30 碰撞能量不足所致。綜合來看,deepScore-α 具有較好的泛化性能,利用Humabody 數據集訓練的模型最終在ProteomeTools2 上的打分效果基本與原數據集上的一致,都優于Comet 和MSGF+。同樣的,打分算法的Top1 的命中率也是其性能評價的一個重要指標。本文也評估了deepScore-α 在ProteomeTools2 數據上的Top1 命中率,實驗結果如表3所示,deepScore-α 在ProteomeTools2數據集上的Top1 命中率相較于另外兩個鑒定工具均提高了5 個百分點,Top1的鑒定結果中Decoy 的數量與另外兩個鑒定工具相比減少了60%左右,因此可以判斷deepScore-α 在打分性能上優于Comet和MSGF+,且具有較好的泛化性能。

圖8 deepScore-α、Comet和MSGF+在碰撞能量為30的Proteome Tools2數據集上的FDR曲線Fig.8 FDR curves of deepScore-α,Comet and MSGF+on Proteome Tools2 dataset with NCE of 30
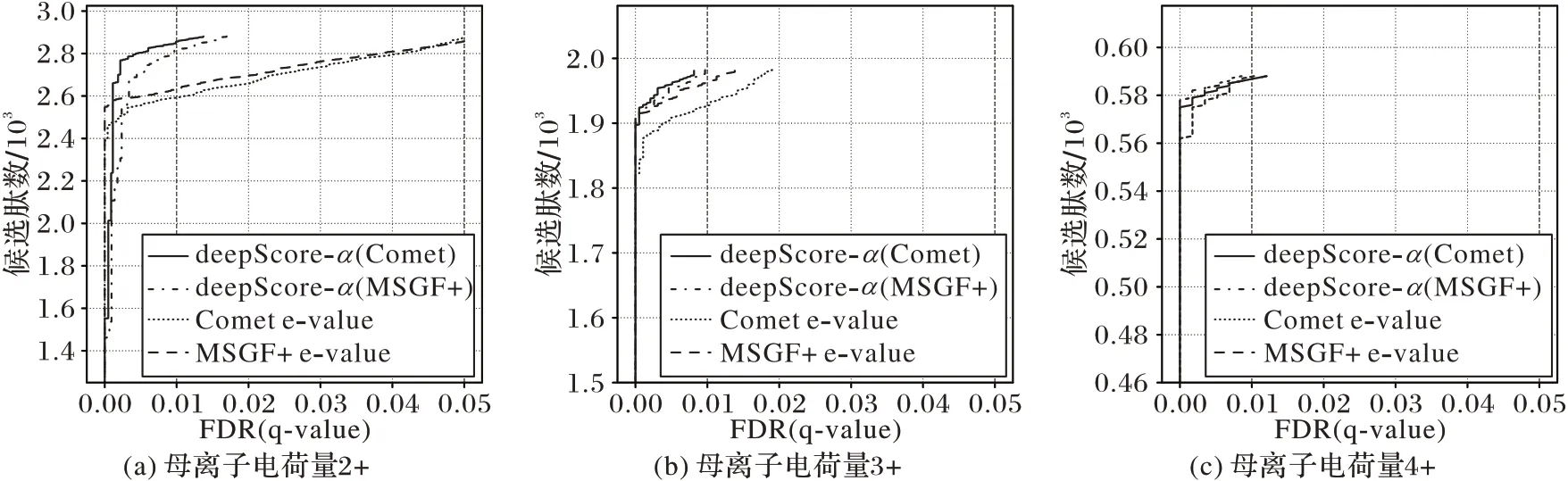
圖9 deepScore-α、Comet和MSGF+在碰撞能量為35的Proteome Tools2數據集上的FDR曲線Fig.9 FDR curves of deepScore-α,Comet and MSGF+on Proteome Tools2 dataset with NCE of 35
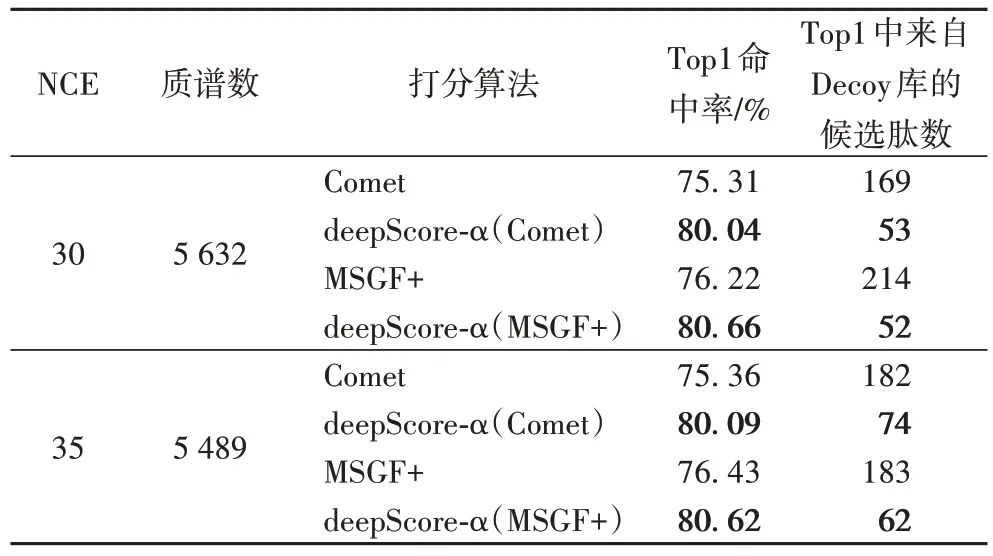
表3 deepScore-α、Comet和MSGF+在ProteomeTools2中打分結果比較Tab.3 Comparison of deepScore-α,Comet and MSGF+scoring results in ProteomeTools2
3 結語
deepScore-α 通過深度學習模型學習肽碎裂規律預測出碎片離子離散化相對強度分布概率,結合實際的碎片離子離散化相對強度形成肽譜匹配得分,有效地利用了深度學習的優勢。deepScore-α 在人類蛋白組數據集上的表現優于常用開源鑒定工具Comet 和MSGF+,且使用人類蛋白組數據集訓練的模型最終在ProteomeTools2 上的表現也優于另外兩個鑒定工具,均可以證明deepScore-α 是一個打分效果優良且泛化能力較強的深度學習打分算法。下一步研究重點是拓展使用的碎片離子范圍,本文在deepScore-α 打分算法中只使用了一價和二價的b/y離子,雖然在肽序列碎裂后產生的碎片離子中b/y離子占大多數,但是其產生諸如a/x、c/z離子以及相應的內部離子是否對打分算法產生影響還需要進一步驗證。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網絡安全與數據管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:36
成都醫學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數學備考)(2020年9期)2021-01-04 00:25:14
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
