以腹瀉為首發表現的糖原累積病Ⅸa型1例并文獻復習
2020-07-04 03:03:59王彩紅卓志強俞蕙
中國現代醫生 2020年12期
王彩紅 卓志強 俞蕙

[摘要] 糖原累積病(glycogen storage disease,GSD)是一組由于先天性酶缺陷所造成的代謝性疾病,其共同特征是糖原代謝異常,導致糖原在肝臟、肌肉、腎臟等組織中貯積量增加。根據缺陷酶及發現的年代順序糖原累積病分為12型。通過分析1例以“間斷腹瀉2個月,發現肝大伴肝功能異常18 d”起病的糖原累積病Ⅸa型患兒的臨床資料和基因特征,總結目前所報道我國的病例相關特點,并進行文獻復習提高臨床醫師對GSDⅨa型的認識。
[關鍵詞] 腹瀉;首發表現;糖原累積病;GSDⅨa型;病例報告
[中圖分類號] R725.8 ? ? ? ? ?[文獻標識碼] C ? ? ? ? ?[文章編號] 1673-9701(2020)12-0166-04
糖原累積病(GSD)是一組由于先天性酶缺陷所造成的代謝性疾病,其共同特征是糖原代謝異常,導致糖原在肝臟、肌肉、腎臟等組織中貯積量增加。根據缺陷酶不同及發現的年代順序糖原累積病分12型[1]。其中GSD Ⅸ是由于糖原磷酸化酶激酶(PHK)缺陷所致,也稱磷酸化酶激酶缺乏癥[2]。GSDⅨ的總發病率1:100000,占所有類型GSD的約25%[3],而GSD Ⅸa最常見,約占總GSDⅨ病例的75%[3]。GSDⅨa系PHKA2基因異常引起,PHKA2基因位于Xp22.2-22.1,包含33個外顯子[4],到目前為止,人類基因突變數據庫(HGMD)報道了與GSD Ⅸa相關的PHKA2基因的110多個突變,包括無義、錯義、剪接、小插入和缺失[3]。GSD Ⅸa的患兒通常表現為肝腫大、肝酶升高和不同嚴重程度的低血糖、血清甘油三酯和膽固醇的輕度升高、生長遲緩、部分有餐后乳酸的升高[3]。本文回顧1例以腹瀉為首發表現的GSD Ⅸa患兒并復習已報道的我國患兒的基因型的文獻,現報道如下。
1 臨床資料
1.1 一般資料
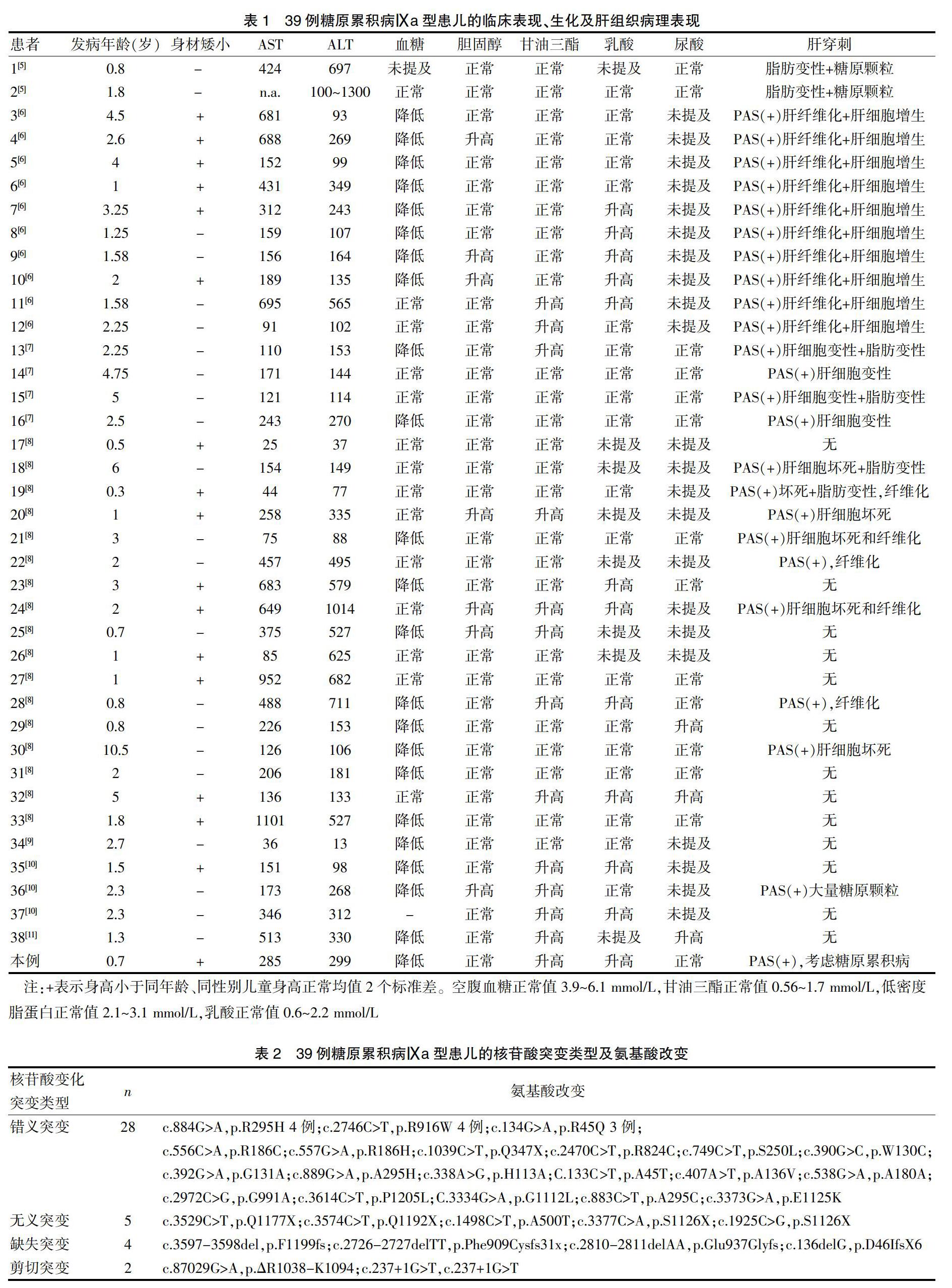
患者,男性,10個月17天,因“間斷腹瀉2個月,發現肝大伴肝功能異常18 d”于2018年7月24日收治入院。患兒為第1胎第1產,足月順產,出生體質量2420 g,智力發育與同年齡兒童相仿,患兒父母非近親結婚。患兒生后無暈厥、無嘔吐、無抽搐等病史,進食次數及量與正常兒童相仿,但2個月前出現間斷腹瀉,大便1~10余次不等,呈稀糊狀,無黏液及膿血便,6月齡至今體重無增長。查體:體重7 kg( 1.2 輔助檢查 ①血常規:WBC 8.5×109/L,RBC 4.18×1012/L,PLT 478×109/L,Hb 116 g/L,N 2.08×109/L。尿常規+尿沉渣:正常。血氣:pH 7.285,Lac 4.3mmol/L,余正常。血氨:73.0 μmol/L,生化全套:ALT 299 U/L,AST 285 U/L,β-羥丁酸0.62 mmol/L,甘油三酯為5.22 mmoL/L,尿酸為172 μmol/L,空腹血糖為2.8 mmol/L↓;肌酸激酶同工酶49.1 IU/L。肝炎病毒均陰性。torch(-)。腹部彩超示肝右肋下7.2 cm、劍突下5 cm、脾未探及腫大;右腎盂增寬,左腎未見局灶性占位,雙側腎上腺區未見明顯局灶性占位。腹水(-)。 1.3 肝組織病理結果 肝細胞彌漫胞漿淡染,胞膜清晰,呈窗格樣改變,少數肝細胞內見大小不一的空泡;肝血竇內枯否細胞未見明顯異常;匯管區纖維組織增生并分隔肝小葉,少量淋巴細胞浸潤。IHC18-2269:CD138(-)、CD31(+)、CD34(血管+)、CD4(+)、CD45(+)、CD68(枯否細胞+)、CD8(個別+)、CK19(膽小管+)、CK7(膽小管+)、EBV(-)、EMA(+)、HBcAg(-)、HBsAg(-)、Hepo(部分+)、Vim(+)、CEA(+)、PAS(肝細胞+)、D-PAS(-)、鐵染色(-)、銅染色(-)、MASSON染色示匯管區纖維組織增生并分隔肝小葉;網狀纖維染色示網狀支架結構存在。考慮遺傳代謝性疾病,首先考慮糖原累積病(圖2)。 1.4 基因分析 全外顯子組檢查結果提示:基因PHKA2,染色體位置chrX:18912486,基因突變信息:NM-000292:exon32:c.3373 G>A(p.E1125K),Hemi,X連鎖隱性遺傳。 1.5 診斷 GSDⅨa型。 1.6 治療及隨訪 給予復方甘草酸苷護肝及生玉米淀粉2 g/kg口服,監測患兒血糖,治療后患兒血糖均在正常范圍,于7月27日出院。出院后繼續予生玉米淀粉2 g/kg口服,患兒出院后2個月至今定期門診進行隨訪,2019年5月25日身高78 cm,體重9.3 kg,無腹瀉,大便1次/d,復查肝功能:ALT 123 U/L,AST 119 U/L,余正常范圍。腹部B超提示肝肋下3 cm、劍突下2 cm。目前患兒病情平穩,繼續隨訪中。 2文獻復習 以“糖原累積病Ⅸa型”“glycogen storage disease type Ⅸa”“PHKA2”為檢索詞檢索中國知網(CNKI)、萬方數據、PubMed等數據庫,檢索時間為2009年8月~2019年8月的文獻,復習相關文獻,回顧性分析已報道我國的具有完整的臨床資料及基因突變資料的GSDⅨa型患兒39例,見表1及表2。 3 討論 GSD Ⅸ是由于PHK缺陷所致,PHK在調節葡萄糖從糖原釋放中起關鍵作用,其缺陷可導致糖原儲存過量[2]。PHK是由四個亞基組成的十六聚酶:α、β、γ和δ。每個亞基都具有其特異性,α和β亞基具有調節功能,γ亞基有催化功能,δ亞基有Ca2+結合功能。α亞基有兩種基因編碼形式:PHKA1和PHKA2,PHKA1突變導致GSD Ⅸd(OMIM 300559),PHKA2突變導致GSD Ⅸa(OMIM 306000)。PHKA1和PHKA2缺陷均為X連鎖隱性遺傳。β和γ亞基分別編碼PHKB和PHKG2,PHKB和PHKG2缺陷為常染色體隱性遺傳[3,4]。 我國已報道的39例GSDⅨa均為男性[5-12],符合X連鎖隱性遺傳的特點。到目前為止,已經通過HGMD記錄了大約119種PHKA2基因突變的患者。目前,有32種突變在中國患者已報道[5-12],包括28個錯義突變,5個無義突變,4個缺失所致的移碼突變,2個剪切突變見表2。外顯子分布在:2(6例)、4(4例)、6(3例)、8(1例)、9(6例)、10(1例)、15(1例)、18(1例)、22(1例)、25(5例)、26(1例)、27(1例)、30(1例)、31(1例)、32(3例),其中53.8%主要集中在外顯子2、4、9、25上,說明外顯子2、4、9、25是中國患者的熱點。 GSD Ⅸa多在嬰幼兒起病,肝腫大和肝酶升高是患者最常見的臨床特征[13,14]。39例患兒多以嬰幼兒起病,發病年齡0.3~10歲,中位年齡為2.39歲。主要表現為肝腫大、肝功能異常,39例患者中肝腫大36例(92.3%),肝酶升高38例(97.4%),低血糖23例(58.9%),身材矮小17例(43.5%),高甘油三酯13例(33.3%)、高膽固醇6例(15.4%),高乳酸12例(30.8%),高尿酸3例(7.7%),1例有運動發育的延遲,無肝硬化和肝腺瘤及肝功能衰竭的報道[13]。這些臨床和生化表現隨著年齡的增加而改善,與之前的報告一致[13]。其發病機制是肝糖原不能及時分解供能,引起低血糖,糖異生功能代償性加強,肝糖原長期在肝臟貯積,使肝臟增大,肝功能異常,并影響生長發育。無低血糖患兒考慮可能糖異生途徑未受到影響;高乳酸血癥、高甘油血癥和高膽固醇的發生率低于50%,可能系患者的酶活性未完全喪失所致。本例患兒以間斷腹瀉為首發表現,考慮可能為淀粉的不完全吸收和影響葡萄糖的吸收有關,具體機制尚不清楚。 39例患兒中26例行肝活檢,主要病理表現為脂肪變性,肝細胞內糖原凝聚,炎癥,壞死,無膽汁淤積及肝硬化。24例PAS染色為陽性,6例脂肪變性,15例肝纖維化,與其他國家有肝硬化患者報告略有不同,可能與種族或基因差異有關系。 GSD Ⅸa既往被認為是一種良性疾病,不需要治療。Zhang J[9]在中國17例GSD Ⅸa患者的隨訪中5例停服生玉米淀粉,隨訪1~10年患者的實驗室數據仍正常。但有肝纖維化、肝硬化的病例報道及其他罕見的PHKA2致病變異導致的認知及語言延遲的表型,最近的研究還表明,一些未經治療的兒童可能會因成長延遲而產生心理困擾及成年骨折風險升高[11,14,15]。而多數學者認為服用生玉米淀粉,不但改善患者的生長速度,使肝功能恢復正常,而且改善患者的肝腫大及肝纖維化[6-8,13]。有研究者報道GSDⅨa服用生玉米淀粉的病例發生缺鐵性貧血,給予補鐵治療能好轉[6]。由于樣本量及隨訪時間有限,生玉米淀粉是否對GSDⅨa的患者至關重要的問題仍有待進一步研究[6]。 綜上所述,對于肝腫大和肝酶升高的患兒,臨床診斷時應注意考慮到GSDⅨa型,基因檢測能夠盡早幫助明確診斷。因部分患兒肝臟病理檢查有發現肝纖維化、肝硬化,提示其結局可能并非良性[13]。早期服用生玉米淀粉能夠改善患者的預后[6,7,14,15]。 [參考文獻] [1] 江載芳,申昆玲,沈穎.諸福棠實用兒科學[M]. 第8版.北京:人民衛生出版社,2015:2262-2263. [2] Bali Deeksha S. Clinical and molecular variability in patients with PHKA2 variants and liver phosphorylase b kinase deficiency[J]. Jimd Reports,2017,37:63-72. [3] Beauchamp NJ,Dalton A,Ramaswami U,et al. Glycogen storage disease type Ⅸ:High variability in clinical phenotype[J]. Molecular Genetics and Metabolism,2007,92:88-99. [4] Achouitar S,Goldstein JL,Mohamed M,et al. Common mutation in the PHKA2 gene with variable phenotype in patients with liver phosphorylase b kinase deficiency[J]. Molecular Genetics & Metabolism,2011,104(4):691-694. [5] Lau CK,Hui J,Fong FNY,et al. Novel mutations in PHKA2 gene in glycogen storage disease type Ⅸ patients from Hong Kong,China[J]. Molecular Genetics & Meta-bolism,2011,102(2):222-225. [6] 王璞,董漪,徐志強,等. 糖原累積癥Ⅸ型12例臨床、病理特點及基因突變位點分析[J]. 肝臟,2018,23(9):14-18. [7] 劉杰,張梅紅,龔敬宇,等. 糖原累積病Ⅵ型和Ⅸa型7例病例報告并文獻復習[J]. 中國循證兒科雜,2017(4):49-53. [8] 張梅虹,王慧君,張萍,等. 磷酸化酶激酶α亞單位新突變所致的糖原累積癥Ⅸ型及核心家系研究[J]. 中華肝臟病雜志,2017. [9] Zhang J,Yuan Y,Ma M,et al. Clinical and genetic characteristics of 17 Chinese patients with glycogen storage disease type IXa[J]. Gene,2017,627:149-156. [10] Fu J,Wang T,Xiao X. A novel PHKA2 mutation in a Chinese child with glycogen storage disease type Ⅸa:A case report and literature review[J]. BMC Medical Genetics,2019,20(1):56. [11] Fengxia Yang,Yi Xu,Chunxiao Fang,et al. Clinical and genetic characteristics of three Chinese patients with glycogen storage disease type Ⅸa Pediatrics and Neonatology[J].Pediatrics and Neonatology,2019,463:466. [12] 郭紅梅,鄭必霞,李玫. X連鎖遺傳糖原累積病Ⅸa一例[J]. 中華兒科雜志,2017,55(5):392-393. [13] Schippers HM,Smit GPA,Rake JP,et al. Characteristic growth pattern in male X-linked phosphorylase-b kinase deficiency(GSD Ⅸ)[J]. Journal of Inherited Metabolic Disease,2003,26(1):43-47. [14] Tsilianidis LA,Fiske LM,Siegel S,et al. Aggressive therapy improves cirrhosis in glycogen:Torage disease type Ⅸ[J]. Molecular Genetics and Metabolism,2013,109(2):179-182. [15] Kim JA,Kim JH,Lee BH,et al. Clinical,biochemical,and genetic characterization of glycogen storage type Ⅸ in a child with asymptomatic hepatomegaly[J]. Pediatric Gastroenterology Hepatology & Nutrition,2015,18(2):138-143. (收稿日期:2020-01-07)
