
電位法測定高含硫水中氯化物的前處理方法的篩選
2020-08-11 02:23:04郝新煥
全面腐蝕控制 2020年2期
關鍵詞:方法
郝新煥
(中國石油獨山子石化分公司研究院,新疆 獨山子 833699)
0 引言
原油中的無機氯和有機氯經過水解或分解作用,在一次和二次加工裝置的低溫部位遇水形成鹽酸復合腐蝕環境,促進金屬腐蝕,造成碳鋼的全面腐蝕,不銹鋼材質的孔蝕和應力腐蝕開裂等,從而造成低溫部位的嚴重腐蝕,嚴重影響著設備的安全運行,因此需要對設備介質中的氯化物進行分析、監測。
但是在實際監測過程中,對于煉油裝置的催化、焦化、加氫裂化等裝置的冷凝水樣,由于水樣性質的復雜性,里面含有大量的硫化物、氨氮,特別是加氫裂化和大煉油的幾套加氫裝置冷凝水中的硫化物含量高達幾千甚至幾十萬mg/L,對氯化物的測定造成嚴重的干擾。
而目前我們所用的測定水中氯化物的方法是GB 11896-89《氯化物的測定 硝酸銀滴定法》,資料和實際情況均顯示硫化物、氨氮對氯化物的測定有嚴重干擾[1],可通過用過氧化氫處理予以消除,但簡單地按照目前方法中含硫化物的前處理方法[2]:加氫氧化鈉溶液將水樣調至中性或弱堿性,加入1mL,30%過氧化氫,搖勻,1min后加熱至70~80℃,已除去過量的過氧化氫,處理硫化物含量在幾十到上萬的水樣并不能夠解決無法檢測的實際問題,因此我們通過大量的試驗評比,進行了長時間的篩選,找到了合適的前處理方法。
1 前處理方法的篩選:
根據我們目前的實際情況:煉油廠催化、重整、蒸餾、焦化、200萬噸加氫、300萬噸加氫、二聯合裝置的監測17個冷凝水樣, 100%含有硫化物,硫化物含量在500mg/L以上的占58.82%,硫化物含量在1000mg/L以上的占52.94%,均多少含有機物,渾濁的水樣占70%左右,對氯化物的測定產生嚴重的影響。結合資料顯示,大量的硫化物、氨氮的存在對氯化物的測定存在干擾,因此在樣品前處理方面,我們主要針對消除硫化物、氨氮干擾方面進行了前處理方法的篩選,主要篩選出以下四種方法:
方法1:在堿性介質中,緩慢滴加適量30%過氧化氫,緩慢加熱,煮沸至無小氣泡產生。GB/T 3050-2000《無機化工產品中氯化物含量測定的通用方法 電位滴定法》
方法2[3]:取一定體積的水樣于250mL 燒杯中,用1mol/L氫氧化鈉溶液調節水樣至堿性(pH=8~9),若水樣本身呈堿性則不必加氫氧化鈉溶液,置電爐上加熱除去NH3,直到蒸汽不再使濕潤的pH試紙變藍色。再用10%硝酸調節水樣至pH=2~6,繼續加熱除去S2-,直到蒸汽不使濕潤的乙酸鉛試紙變黑。加入足量的30%過氧化氫除去SO32-、S2O32-以及剩余的硫化物等干擾物質,加熱過程中適時補充蒸餾水以防止燒干;
方法3[4]:用中速濾紙將樣品過濾后,移取濾液10~100mL于250mL的三角燒瓶中, 加入蒸餾水使總體積約100mL和兩粒玻璃珠;將三角燒瓶置于電爐上加熱, 沸騰后微沸7min以上(溶液變成乳白色或無色);用1mol/L HNO3滴加溶液至pH2~3(用玻璃棒拈此溶液于pH試紙上觀察), 微沸2~3min,接著加入2mL,30%過氧化氫,繼續微沸2~3min;加1mol/L NaOH 溶液,使溶液呈堿性(用玻璃棒拈此溶液于pH 試紙上觀察pH≥9);繼續微沸5~10min取下,此時溶液無色透明,(中途若水蒸發過多,可補加一點蒸餾水);水冷至室溫后, 往溶液中添蒸餾水至約50mL;
方法4:移取適量的水樣于250mL燒杯中稀釋至50mL,用1mol/L的氫氧化鈉溶液調節pH值<12,加入10mL過氧化氫,然后再用1mol/L的氫氧化鈉溶液調節pH值<12。把裝有試樣的燒杯放在電熱板上微沸防止溶液濺失,并不斷地加入1mol/L的氫氧化鈉溶液保持樣品呈堿性,直到把水樣中過量的過氧化氫除盡并保證溶液仍在堿性條件下,煮沸過程中要不斷加入蒸餾水防止煮干。Q/SY DS 01 J.506-2009《煉油廠廢水中氯化物含量測定-電位滴定法》。
1.1 試驗1
針對前面提出的前處理方法,考慮到樣品的復雜性,出現問題不易查找,我們首先采用標液,氯化物標液:10mg/L;取樣量:50mL,采用不同的前處理方法處理后,用自動電位滴定儀逐一進行了測定,結果如表1所示。
從表1試驗結果來看,平行結果稍好的是方法1和方法2,說明這兩種方法是可行的。
1.2 試驗2
針對方法1和方法2用實際樣品進行了檢測,考慮到樣品中氯化物的含量可能較低,在檢出限以下,對測定結果會有影響,因此在實際樣品中加標進行了進一步試驗:
樣品取樣量為20mL,加標Cl-為100mg/L,加入量為4mL,分別采用方法1和方法2,測定結果如表2所示。
從表2試驗結果可以看出,方法2的平行結果較好是,方法2是可行的。
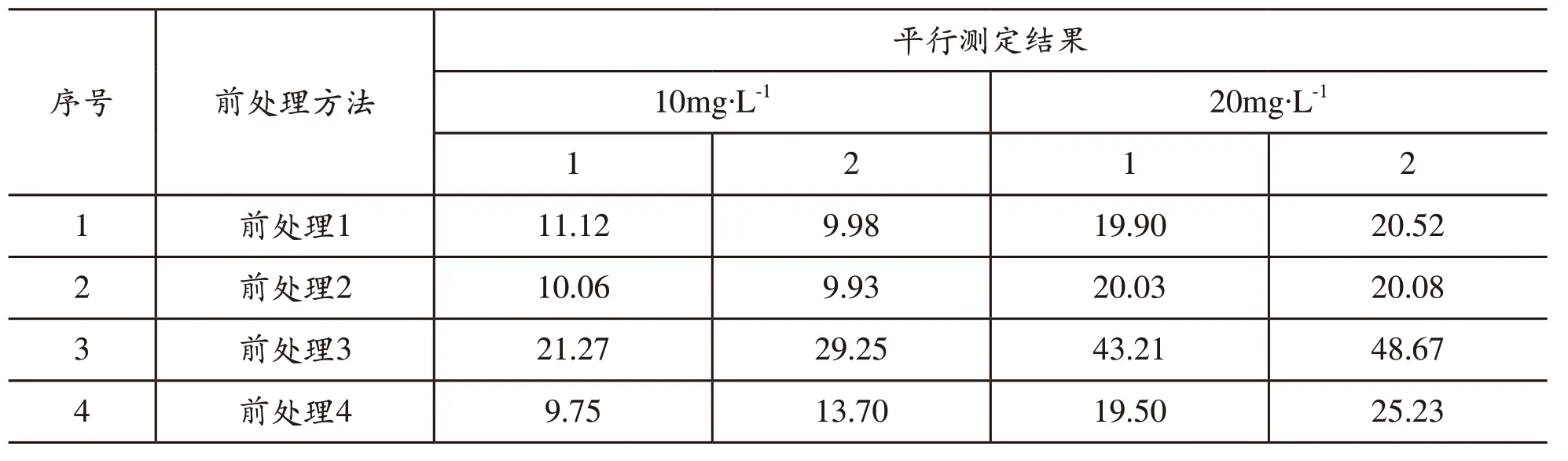
表1 前處理方法篩選試驗1

表2 前處理方法篩選試驗2

表3 精密度試驗

續(表3)

表4 加標回收率試驗
2 評價
對選出的前處理方法,我們又通過對實際樣品進行多次的精密度和加標回收率試驗進行了驗證。
2.1 精密度
如表3所示。
2.2 準確度即加標回收率
如表4所示。
3 結論
通過大量的試驗,我們找出了合適的電位滴定法測定高含硫污水中氯化物的前處理方法,并通過對實際樣品進行多次的方法精密度和準確度的評價,精密度的相對標準偏差最大為1.96%,實際樣品的加標回收率在99.32%~104.73%,確定此前處理方法的可靠性。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
意林原創版(2016年10期)2016-11-25 10:28:30
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
