STXBP1相關發育性和癲癇性腦病1例及文獻綜述
2020-08-12 06:06:40劉維亮何志旭
山西醫科大學學報 2020年7期
劉維亮,李 芳,何志旭,艾 戎
(1 貴州醫科大學附屬醫院兒科,貴陽 550004;2 貴州醫科大學附屬醫院眼科;*通訊作者,E-mail:liuweiliang205@aliyun.com)
癲癇性腦病是一種嚴重腦部進行性疾病,以頻繁或嚴重的癲癇發作以及大量的發作間期陣發性異常腦電活動為主,可導致進行性認知和行為障礙[1]。既往在大田原綜合征和幾種早發性癲癇腦病患者中發現有突觸融合蛋白結合蛋白1(syntaxin-binding protein 1,STXBP1)基因突變[2]。這些患者多在嬰兒早期發病,有難治性叢集性運動性驚厥發作。并且在腦電圖上多表現為發作間期爆發-抑制模式。腦MRI多為正常。預后差,多伴有嚴重的精神運動障礙,嬰兒期死亡率高。存活患者通常表現出明顯年齡依賴性癲癇演變特性[3,4]。另外在遲發性嬰兒痙攣伴部分呼吸鏈復合物Ⅳ型缺乏癥、無癲癇病史的非綜合征性智力障礙和Dravet綜合征等患者中也發現了該基因突變[5-7]。STXBP1基因突變患者具有高度的臨床異質性,隨著世界范圍內對該類疾病認識的提高,患者智力損害不僅與癲癇相關,也與遺傳發育性異常有關,國內雖然報道過STXBP1基因突變癲癇患者,但仍然診斷為癲癇性腦病[8]。我們報告了1例STXBP1基因突變癲癇患者,在國內首次診斷其為STXBP1相關發育性和癲癇性腦病患者,國內首次明確該病診斷概念,并總結STXBP1基因突變相關患者臨床特征,以期在表型上能更全面地認識該類疾病。
1 對象與方法
1.1 研究對象
患者,男,9月,因“3 d內抽搐13次”入院,并有左側陰囊較對側大。抽搐前智力運動正常,抽搐后智力運動倒退至新生兒水平,入院診斷:①癲癇;②癲癇性腦病;③左側腹股溝斜疝。該患者及其父母為本研究對象。我們總結了國外與該患者有相同基因突變位點患者的臨床特征和治療經驗,且總結了國內外與該患者有相同致病基因的病例特點。
1.2 研究方法
1.2.1 基因組DNA的提取 留取患者和其父母外周靜脈血3 ml,EDTA抗凝,酚-氯仿法提取基因組DNA,于-20 ℃冰箱保存備用。本研究得到了貴州醫科大學附屬醫院倫理委員會的批準,基因分析獲取了患者家長的知情同意。
1.2.2 基因測序和驗證 應用北京康旭醫學檢驗所二代測序進行患者及其父母三人全外顯子檢測,應用Cavoris儀打斷基因組約200 bp,使用XGen Exome Research Panel v1.0工具包(Integrated DNA Technologies,Skokie,Illinois,USA)捕獲外顯子,外顯子的所有長度為35.58 Mb。然后在Illumina Hiseq2500平臺(Illumina,San Diego,CA,USA)標準化上機測序樣本。測序結果用Burrows-Wheeler比對儀(BWA)與人類參考基因組(GRCH37/HG19)進行比對。用Samtools和Pindel軟件分析了單核苷酸變異和序列的插入和缺失,在相關的變異頻率數據庫(ESP,dbSNP,1000Genomes,ClinVar和HGMD)中檢查所選的變異,應用致病性預測工具(Polyphen 2,SIFT,Mutation Taster)進行預測。針對突變位點應用Sanger測序法進行一代測序驗證,基因序列分析應用DNAstar軟件進行序列分析和比對。
2 結果
2.1 臨床表現及實驗室檢查結果
患者9月齡時因“3 d內抽搐13次”入院,表現為無明顯誘因出現雙眼上斜凝視(左側為主),偶有口唇抽動吐沫,四肢不對稱強直,偶有肢體抖動,意識喪失,持續10-20 s后緩解,白天夜間均有發作,抽后嗜睡。抽搐前智力運動正常,抽搐后智力運動倒退至新生兒水平,頭抬不穩,坐不穩,不能站。一直不會發聲。抽后不會笑,不認人。出生史:第2胎第2產,孕36+6周因母親患高膽汁酸血癥剖宮產,出生體質量2.6 kg,出生時無特殊。既往史及家族史均無特殊。有一3歲6月哥哥,體健。
入院查體:體溫37.1 ℃,血壓95/49 mmHg,呼吸28次/min,脈搏118次/min,身高75 cm,體質量9 kg,頭圍45 cm,前囟約1.0 cm×1.0 cm,平軟,張力可;神清,頭顱五官無畸形,全身皮膚未見皮疹,淺表淋巴結未捫及,雙瞳孔等圓等大,直徑3 mm,對光反射靈敏,眼球運動正常,口腔黏膜光滑,心肺腹神經系統查體無特殊,枕部平坦,左側陰囊較對側大。
輔查結果:血乳酸2.27 mmol/L(0.6-2.2 mmol/L),β-羥基丁酸0.94 mmol/L(0.03-0.3 mmol/L),血常規、肝腎功電解質、血脂、同型半胱氨酸、血氨、銅藍蛋白、維生素D和B12、葉酸、骨源性堿性磷酸酶、C反應蛋白、凝血均無異常。腦脊液常規、生化、墨汁染色、革氏染色、抗酸染色、細菌培養無異常。心電圖:竇性心律不齊。心臟多普勒超聲正常。顱腦MR:雙側海馬體積縮小,雙側側腦室顳角增寬,雙側顳葉內側腦溝增寬,左側明顯。雙側海馬MRS(見圖1):左側氮-乙酰天門冬氨酸/(膽堿+肌酸)[NAA/(Cho+Cr)]比值0.46,右側NAA/(Cho+Cr)比值0.53(正常參考值>0.72),提示雙側海馬硬化。
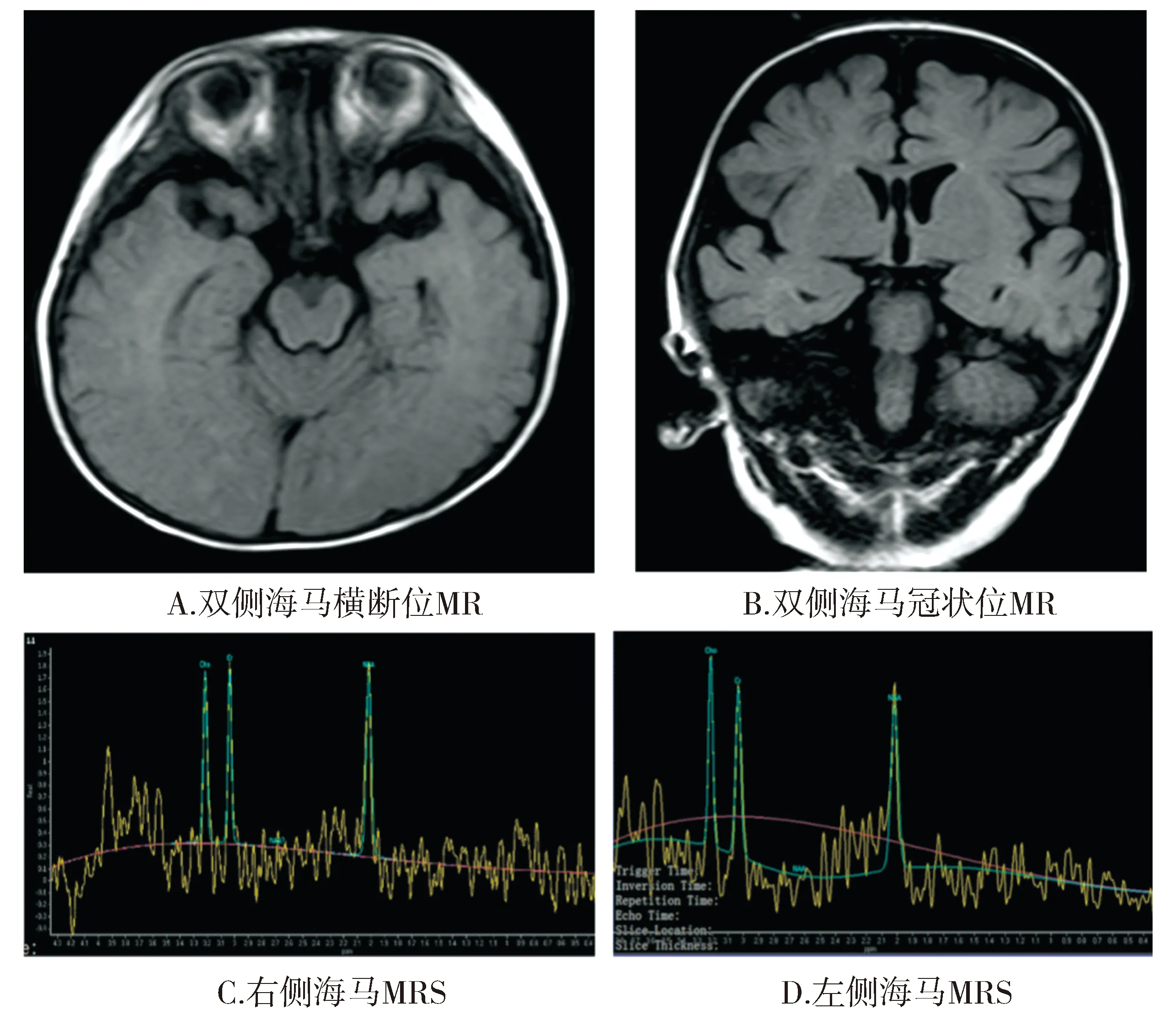
圖1 患者腦MR和雙側海馬MRS
入院予丙戊酸24 mg/(kg·d)治療,抽搐發作部分好轉;入院1周內給予咪達唑侖1-0.5 μg/(kg·min),抽搐發作控制,逐漸減停咪達唑侖抽搐部分好轉。入院10 d查視頻腦電圖(VEEG):監測期間無特殊臨床事件發生,醒睡各期雙側各導彌漫性2-3 Hz中-高波幅慢波發放,背景彌漫性慢波發放,無枕區優勢節律(見圖2A)。入院半月后,逐漸予托吡酯6.94 mg/(kg·d)治療,對抽搐發作無明顯效果。入院1月后,加用氯硝西泮后發作明顯減少,氯硝西泮用量為0.2-0.04 mg/(kg·d),因足量使用后明顯嗜睡,導致吸入性肺炎減量,偶有每天雙眼向左凝視半分鐘。入院39 d復查VEEG:監測期間無特殊臨床事件發生,背景慢化好轉,醒睡各期雙側頂、枕、后顳大量1-2 Hz中-高波幅慢波陣發,慢波主要局限在后頭部枕后顳區,前額中央頂區慢波明顯好轉(見圖2B)。入院2月(11月齡)后逐漸予左乙拉西坦30 mg/(kg·d)后抽搐停止。1歲3月復查VEEG正常(見圖2C)。1歲3月起可認人,會笑,生氣時易雙手抖動,無肢體松軟,可獨坐,可扶站不會走,不會咀嚼僅能吞咽,易過度興奮和嗜睡交替。現1歲10月僅左乙拉西坦治療癲癇,近11月無癲癇發作,現反應能力差,眼神交流差,不會發音,無雙手抖,無興奮思睡,可扶站不會走,咀嚼差。
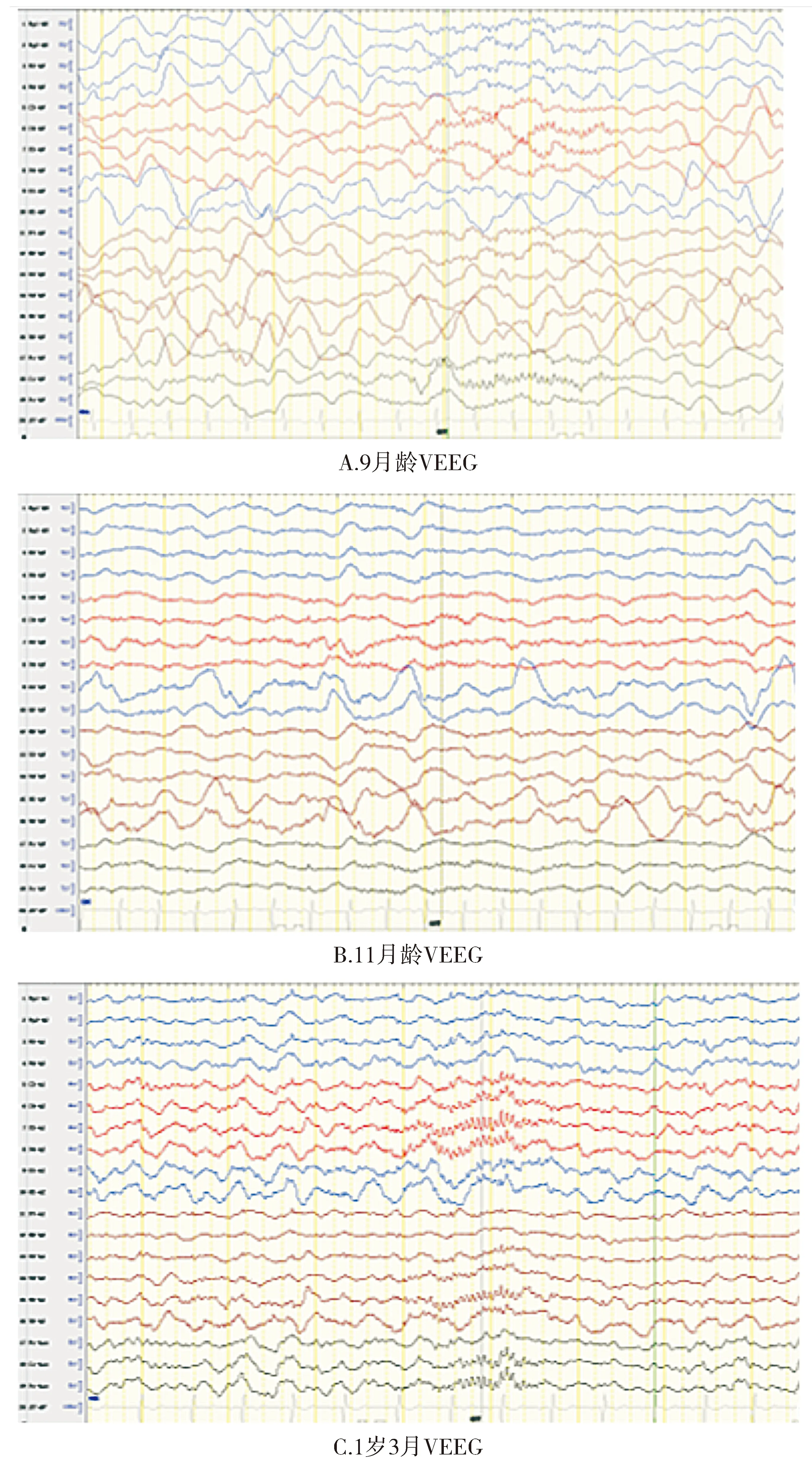
圖2 患者視頻腦電圖(VEEG)
2.2 基因檢測結果
本例患者基因分析證實在STXBP1基因第18外顯子中第1651位核苷酸C突變為T(c.1651C>T,見圖3A),氨基酸變化為p.A rg551Cys(p.R551C),患者父母親未發現該位點核苷酸變異,證明患者為新發突變,患者父母親基因型正常(見圖3B和3C)。患者突變位點氨基酸為高度保守位點(見圖4)。該突變位點既往國外有報道[9-11]。根據ACMG指南(2015年)[12],通過PS2+PS1+PM1+PM2+PP3致病證據證明患者變異為致病性突變。
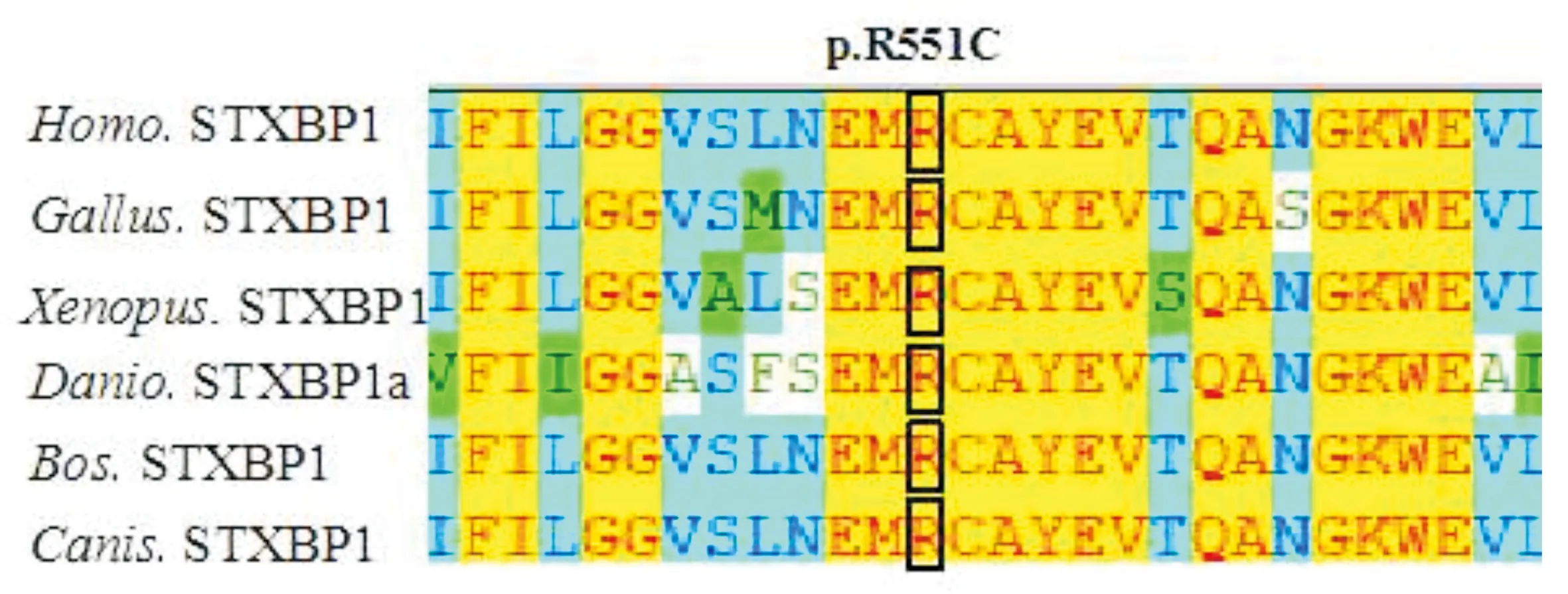
圖4 患者STXBP1 p.R551C突變氨基酸位點保守性分析
2.3 STXBP1基因突變患者的臨床特征
結合患者的臨床特征,基因突變檢查結果,本例報道的患者診斷為癲癇,STXBP1相關發育性和癲癇性腦病,左側腹股溝斜疝。在既往國外報道臨床資料較全的2例患者與我們報道的病例有STXBP1基因相同p.R551C突變的患者中,病例1[9]為男性患者,病例2[10]為女性患者,這些患者均有智力障礙,驚厥發作有痙攣、強直、陣攣、局灶性癲癇發作和失張力發作(見表1)。腦電圖均有背景慢化,無高峰節律紊亂。嬰兒期起病,我們報道的患者起病月齡較晚,治療效果好于國外報道的2例患者。另外國外報道1例STXBP1基因相同p.R551C突變的患者為孤獨癥患者[11]。國外還報道1例STXBP1基因相同位點突變p.R551G(c.1651C>G)的患者是有智力障礙的孤獨癥男性患者[13]。
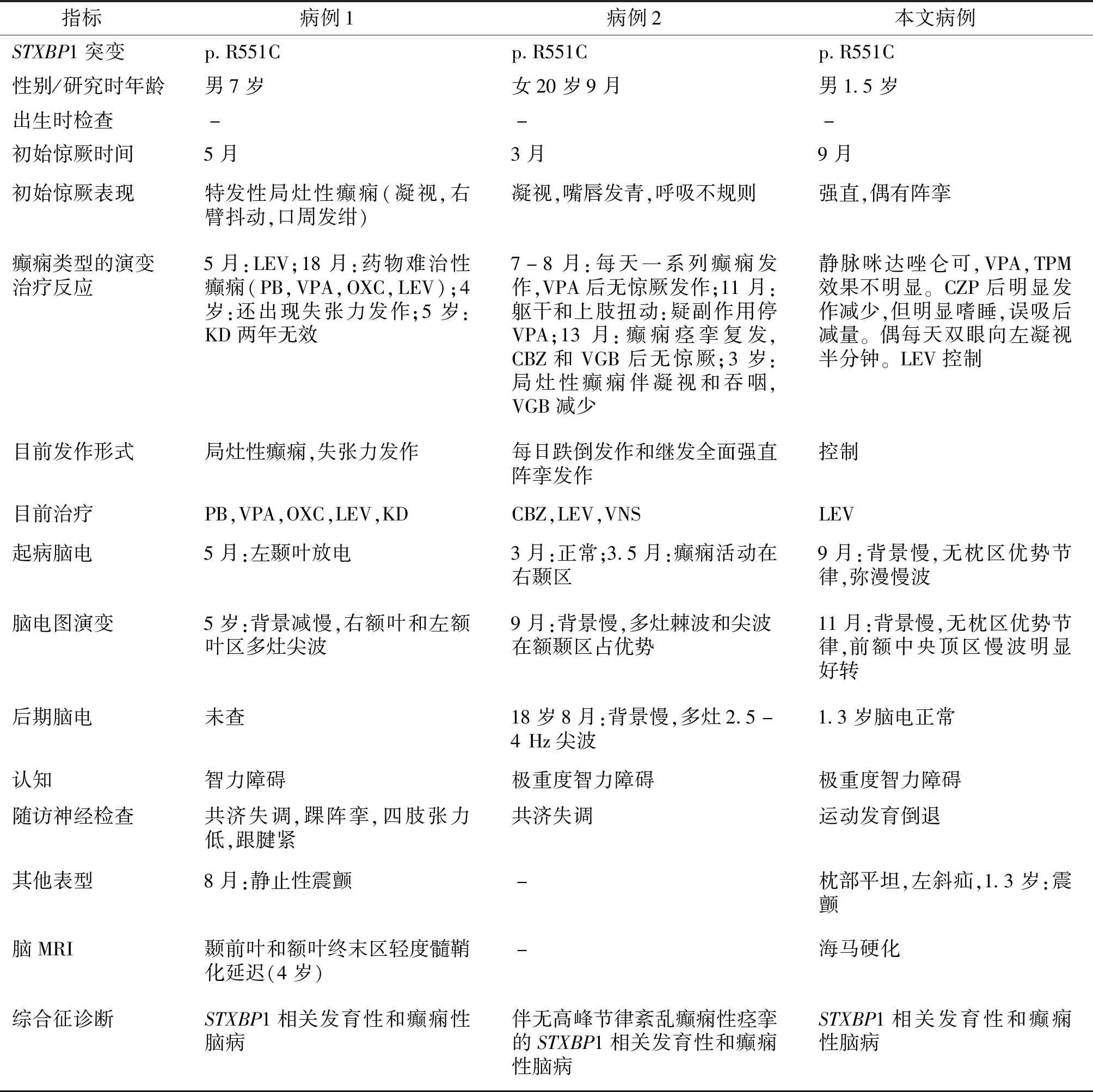
表1 世界范圍內STXBP1 p.R551C突變患者的臨床數據
3 討論
突觸融合蛋白結合蛋白1(STXBP1)主要是在大腦中表達的膜轉運蛋白家族中的一種蛋白,其在神經遞質分泌的突觸囊泡對接和融合中起重要作用。STXBP1基因編碼該蛋白,由20個外顯子組成,位于染色體9q34.11上[14,15]。該基因有錯義、移碼、無義和剪接位點突變及基因內和包括該基因的染色體大片段異常等。STXBP1突變患者的表型譜很廣,國內外報道STXBP1基因突變患者有大田原綜合征、嬰兒痙攣癥、未分類的早發型癲癇和腦病、Dravet綜合征、Rett綜合征、非綜合征性癲癇、智力障礙和孤獨癥等[2-7,11,13,16,17]。表現為廣泛復雜神經發育障礙類疾病,而不是原發性癲癇性腦病,應稱之為STXBP1腦病,包括有癲癇發作和非癲癇發作類疾病[17]。該類患者的臨床特征:①約95%的患者有癲癇,最常見的發作類型是痙攣發作(65.3%)、局灶性發作(57.9%)和強直性發作(41.3%),有1/3以上患者通過治療可無癲癇發作,有1/3患者為難治療癲癇;②腦電圖檢查中有60%以上有局灶性或多灶性癲癇波活動,有35.9%為暴發抑制和40%高峰節律紊亂;③所有患者均有智力障礙,88.4%患者為重度到極重度的智力障礙;④約五分之一的患者中出現孤獨癥或孤獨癥特征的行為問題;⑤常見運動異常特征有軸向低肌張力、共濟失調、意向性震顫、運動障礙或肌張力障礙等;⑥約一半的患者中影像學檢查(腦MRI)正常,其他的異常有腦萎縮(33.3%)、胼胝體變薄(16.2%)、髓鞘發育不良或延遲(16.2%)[17]。
STXBP1腦病多在新生兒到嬰兒期發病,早期癲癇患者最初主要特征是運動性癲癇(局灶性或全身性),初始發作間期腦電圖異常,有正常的頭圍發育。此外,患者經常有中到重度發育遲緩。患者常有共濟失調步態。出現這些特征時臨床醫生應考慮STXBP1基因的分子篩選[18]。
STXBP1腦病中沒有發現癲癇的起病年齡或持續時間與智力嚴重程度相關,患者基因型和表型的關系不大,STXBP1在神經發育的許多方面發揮重要作用,該基因在腦內可通過多個維度干預神經系統發育,該類疾病除了有智力障礙和癲癇外,還有其他多種神經系統異常表現,包括孤獨癥或孤獨癥特征、運動障礙、肌張力障礙、震顫、肌張力低下、共濟失調、錐體外系異常等表現[17]。我們報道的STXBP1基因相同p.R551C突變的患者就有很多表型,故遺傳背景或環境因素等其他因素可能在形成最終表型中起作用。我們報道的患者在嬰兒期發病,在發病早期有頻繁驚厥和智力低下,用藥物控制驚厥發作后腦電圖正常但仍有明顯智力運動發育障礙,其為STXBP1早發性癲癇和腦病,但與早發性癲癇腦病易致概念混淆,我們診斷為STXBP1相關發育性和癲癇性腦病。因2017年國際抗癲癇聯盟重新定義一類新的疾病為發育性和癲癇性腦病的概念,該病主要是由于遺傳因素引起,有早期發病,難治性癲癇發作,發育遲緩或停滯等,發育性異常和癲癇因素均可導致腦病[19],該類腦病可以與致病基因名合并命名[19,20]。且這些患者較易并發智力障礙、孤獨癥譜系障礙(ASD)和行為問題等[21]。我們在國內首次提出診斷該類疾病,以期更好地認識該類疾病,對本病的病因治療可能更有幫助。
STXBP1腦病的治療需要多學科協同治療,目前其癲癇治療主要是癥狀性治療,其行為和運動問題需要運動療法和作業治療,以最大限度地發揮發育潛力,該類疾病的癲癇治療多較困難,且多需要抗癲癇藥聯合應用,其中最常被報道有效的藥物是丙戊酸,其次是左乙拉西坦和氨己烯酸[17]。在一些研究中,氨己烯酸對痙攣發作有很好的反應[16,22,23],丙戊酸[22,23]和左乙拉西坦[24]也可有同樣的效果。在動物模型實驗中,通過VEEG顯示,不同基因組背景的Stxbp1+/-小鼠再現人類所觀察到的癲癇表型,其肌陣攣和棘波放電能被左乙拉西坦抑制。推測左乙拉西坦具有結合到突觸囊泡釋放糖蛋白SV2A的突觸前作用模式,可以抑制小鼠模型中由突觸前基因Stxbp1基因突變引起的腦電異常[25]。有報道生酮飲食成功治療STXBP1腦病的1例癲癇患者,是生后10 d發病的STXBP1基因p.R406C突變的患者,其初始表現為眨眼,嘴唇抽動,眼睛向右側偏離,不對稱強直,后發展為嬰兒痙攣癥,給予生酮飲食聯合托吡酯治療后癲癇發作控制[26]。另外,有文獻報道兩例STXBP1腦病患者接受癲癇外科手術后癲癇治療效果良好,1例在生后5 d發病的STXBP1基因p.gly544val突變患者,其初始發作為肌陣攣發作伴凝視,5個月時出現不對稱屈肌痙攣。對苯巴比妥、卡馬西平和皮質類固醇激素有暫時效應,對氨己烯酸、托吡酯、左乙拉西坦、硝西泮和維生素B6無效,腦MRI正常,8月齡時腦電圖表現為慢波和癲癇波活動,在右枕顳區最顯著,隨后硬膜下網格腦電圖在腦右后區記錄到發作期多發性痙攣發作波形,在1.5歲時患者被實施右側顳葉枕部完全離斷和多處軟腦膜下橫切術,術后右側枕葉活檢示局灶性皮質發育不良1a型,手術后癲癇發作頻率立即降低了95%,父母反應生活質量有明顯改善[10],故推測STXBP1突變與局灶性皮質發育不良可能存在因果關系[10];另一例STXBP1基因p. Thr78His fs*40突變患者為男性試管嬰兒,沒有癲癇家族史,為出生體質量870 g的25周早產兒,接受了9 d的機械通氣治療,此后一直正常,沒有任何并發癥,直到4月齡(糾正)時被發現有智力發育遲滯,在5個月時接受了腦磁共振成像,腦電圖和染色體檢查沒有明顯異常,6月齡時出現了伴腦電圖高峰節律紊亂的嬰兒痙攣癥,予促腎上腺皮質激素治療,僅產生短暫的效果,9月齡仍有明顯痙攣發作和智力運動發育遲滯及皮質盲,腦MRI顯示胼胝體發育不全,腦白質髓鞘形成延遲,雙側額葉中度萎縮,但無腦室周圍白質軟化,甲狀腺素釋放激素,托吡酯、拉莫三嗪對痙攣發作或腦電異常無效,1歲1月時接受了胼胝體切開術治療,術后未再出現癲癇發作和腦電異常,盡管仍不能坐或看,但患者能表現出基本的情感反應[27]。
綜上所述,我們在國內首次診斷STXBP1相關發育性和癲癇性腦病,國內首次明確該病診斷概念。該病患者起病年齡小,進展快,以反復抽搐(痙攣、局灶性發作、強直)為主要表現,智力運動明顯倒退,發育性異常和癲癇因素均可導致腦病,與普通癲癇比較治療效果差,治療方面急性期我們采用左乙拉西坦聯合丙戊酸和小劑量苯二氮類藥物有效,病情平穩后左乙拉西坦維持治療效果好。該類疾病國內外多以左乙拉西坦或其聯合多種抗癲癇藥治療為主[17,24,27],生酮飲食和癲癇外科手術治療在部分個案患者中有一定希望。STXBP1相關發育性和癲癇性腦病屬于STXBP1腦病中最常見的一類疾病,該類疾病屬于復雜神經系統發育異常疾病,病因治療需要進一步研究。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國民間療法(2021年5期)2021-06-09 09:21:04
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
飲食科學(2017年5期)2017-05-20 17:11:53
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
安徽醫科大學學報(2015年9期)2015-12-16 11:09:44
西南軍醫(2015年4期)2015-01-23 01:19:30
中國中醫藥現代遠程教育(2014年20期)2014-03-01 04:31:21
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
