摻雜改性對鈣鋁基復合物酯交換催化劑吸附性能影響的分子模擬
2020-08-19 04:07:46厲志鵬牛勝利韓奎華路春美
化工學報 2020年8期
厲志鵬,牛勝利,韓奎華,路春美
(山東大學能源與動力工程學院,山東濟南250061)
引 言
隨著全球范圍內氣候變暖情況的出現,人們對綠色可持續可再生燃料的關注日益增加。與化石燃料相比較,生物柴油有著無毒、可生物降解、可再生等諸多優勢,并且具有生命周期近碳中性優勢[1]。在商業生產中,以脂肪酸甲酯(FAME)形式存在的生物柴油是使用均相堿催化劑催化甘油三酸酯(植物脂肪和動物油脂的主要成分)與甲醇的酯交換反應生產的。盡管該催化過程在溫和的條件下便能提供很高的反應速率,但甘油三酸酯與甲醇進行酯交換反應時,將產生大量副產物甘油,產物凈化程序復雜,由此增加了生物柴油的生產成本[2],采用乙酸甲酯代替甘油三酸酯與甲醇進行反應,酯交換生成的副產物為甘油三乙酸酯,可完全溶于脂肪酸甲酯,作為燃料生物添加劑成為生物柴油燃料的一部分,同時這也是當前分子模擬普遍采用的方法[3-4]。該生產過程的另一個缺點就是均相催化過程催化劑分離困難,并產生大量廢水,相較而言,非均相催化是一種環境友好的生產生物柴油的有效途徑。
γ-Al2O3具有極高的熱穩定性、高機械穩定性、高比表面積以及較大的孔徑和孔容,作為催化劑載體已經廣泛應用于多種化學反應過程,如聚合[5]、改性[6]、脫水[7]、質子化[8]。另外,鈣基催化劑堿性強,催化酯交換反應條件溫和。目前,已有大量關于單一鈣基[9]及其復合金屬化合物[10]催化甘油三酸酯與甲醇酯交換反應生成生物柴油的相關研究。但是,鈣鋁二元復合氧化物固體堿催化劑仍然存在比表面積較小導致的催化活性弱以及活性位浸出率較高造成的重復使用性差等問題。Zheng 等[11]制備的鈣鋁多相催化劑在6 次循環使用后催化劑的質量從0.150 g 減少到0.075 g,活性位氧化鈣損失較多導致失活。Tang 等[12]在對鈣鋁復合催化劑進行SEM 分析時指出,鈣鋁復合催化劑顆粒較大,團聚現象嚴重,不利于酯交換反應的進行。因此,目前研究的熱點主要集中在提高鈣基催化活性以及降低鈣基活性位流失率等方面。
鋅、鑭、鎂改性可有效強化CaO 的催化酯交換性能。鋅-鈣氧化物共存可以提高催化劑的比表面積和比孔容,形成粒徑小的鈣鋅復合物,提高催化酯交換的活性[13]。La3+擁有完整的4d和空白的5d軌道以提供或接收電子,使催化劑擁有酸堿雙性位,其鍵合作用能有效抑制催化酯交換過程中鈣基活性位溢出,使催化劑具有較高的重復使用性[14]。與純CaO 催化劑相比,鎂改性的鈣鎂復合物催化劑可降低活性位浸出率、提高催化劑穩定性、延長催化劑的使用壽命[15]。相關研究同時得出,分別以Al2O3和CaO 作為催化劑的載體和活性位,進一步進行鋅[16]、鑭[17]或者鎂[18]的摻雜改性,能顯著提高鋁鈣復合物的催化性能,比單純的鈣鋁復合催化劑效果更佳,但其中的作用機制尚不清楚。
本文基于密度泛函理論(density function theory,DFT),對Ca-Al-M(M=Zn,La,Mg)復合物催化劑表面的電子結構特性進行了分子模擬,計算了復合催化劑形成時的摻雜結合能,系統地探究了甲醇在催化劑表面的吸附情況,通過Mulliken 電荷布局分析討論了甲醇在催化劑表面吸附后電荷的變化,在分子層面上解析了鋅、鑭、鎂摻雜改性對鈣鋁復合物催化劑催化酯交換過程的影響機制,所得結論可為鈣鋁基固體堿酯交換催化劑的設計與開發提供理論指導。
1 計算方法和模型
1.1 計算方法
本研究基于密度泛函理論,采用自旋非限制廣義 梯 度(generalized gradient approximation)耦 合Perdew-Wang 91 泛函的方法(GGA-PW91)在DMol3水平上完成[19]。選用有效核心贗勢(DFT semi-core pseudopod)以降低計算成本,同時價電子波函數采用雙數值基加軌道極化函數(DNP)展開,并將Monkhorst-Pack 網格參數k 設置為2×2×1。計算精度為Customized,自洽場最大迭代次數為300,選用熱拖尾效應(smearing),并保持初始默認值0.136 eV。結構優化以梯度、位移和能量是否收斂為判據,最大梯度、最大位移和最大能量的收斂精度均分別優于0.110 eV、5.0×10-4nm 和5.4×10-3eV/nm,同時,SCF收斂判據的截斷值tolerance為1.0×10-5。
本文計算了酯交換反應物甲醇、乙酸甲酯分子的鍵長,并與文獻結果對比以確保優化方法的準確性。甲醇和乙酸甲酯的結構如圖1 所示,計算結果見表1,其中鍵長與文獻[4,20]結果相符,因此本文所用計算方法可行。

表1 甲醇和乙酸甲酯的鍵長Table 1 Bond length of methanol and methyl acetate
1.2 模型構建
本文所用γ-Al2O3結構的晶體數據來自Digne模型[21]。對該結構進行進一步構型優化以完善模型,計算得出γ-Al2O3晶體參數分別為a=0.559 nm、b=0.841 nm、c=0.807 nm、β=9.059 nm,與文獻結果吻合。鑒于(110)表面為γ-Al2O3晶體暴露最豐富表面[22],因此本文選用γ-Al2O3(110)表面作為計算表面。模型采用p(1×1)的7 層周期性平板,并在平板與真空層構成超單元晶胞,同時在z 方向構建1 nm真空區以避免連續平板之間的相互作用,從而在空間中周期性重復,最終得到γ-Al2O3(110)表面模型,如圖2 所示。考慮表面弛豫影響,固定底部四層原子,同時保持上部三層及吸附分子自由移動。γ-Al2O3(110)表面暴露有三配位鋁(Al3c)、四配位鋁(Al4c)、五配位鋁(Al5c)以及二配位氧(O2c)、三配位氧(O3c),其中三配位鋁(Al3c)、四配位鋁(Al4c)、二配位氧(O2c)未飽和,這與Song等[23]的研究結果一致。
氧化鋁表面的摻雜結合能(Edope)(單原子摻雜)由式(1)計算。

氧化鋁表面的摻雜結合能(Edope)(雙原子摻雜)由式(2)計算。
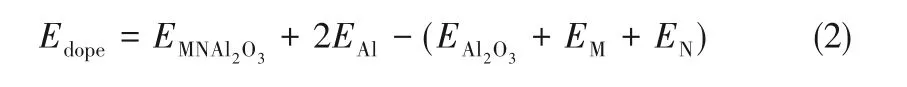
催化劑表面的吸附能(Eads)由式(3)計算。吸附能可以用來衡量吸附體系的穩定性,其值越大表明吸附過程中放出熱量越多,吸附越穩定,催化劑催化效果越好。
2 結果和討論
2.1 Ca-Al-M復合催化劑的構成
在對Ca-M 在氧化鋁表面摻雜過程中,考慮到γ-Al2O3(110)表面有三個不同配位的鋁原子(Al3c-1,

圖1 甲醇結構和乙酸甲酯結構(紅色為氧原子,白色為氫原子,灰色為碳原子)Fig.1 Structure of methanol and methyl acetate(O atoms are red,H atoms are white and C atoms are gray)

圖2 γ-Al2O3(110)結構表面(紅色為氧原子,粉色為鋁原子)Fig.2 Structure of the γ-Al2O3(110)surface(O atoms are red and Al atoms are pink)
Al4c-1,Al5c-1),利用原子替換法[24]分別對氧化鋁進行摻雜,摻雜的類型及優化后摻雜結合能如表2 所示。摻雜結合能為正值表明該摻雜過程不能自發進行,相應的值越小,表明摻雜過程越容易進行,摻雜得到該結構的可能性越大[25]。結果表明,Ca-Al 復合物催化劑中的鈣原子傾向于摻雜在五配位鋁Al5c的位置;Ca-Al-Zn 復合物催化劑中的鈣原子、鋅原子分別傾向于摻雜在四配位鋁Al4c、五配位鋁Al5c的位置;Ca-Al-La 復合物催化劑中的鈣原子、鑭原子分別傾向于摻雜在三配位鋁Al3c、五配位鋁Al5c的位置;Ca-Al-Mg 復合物催化劑中的鈣原子、鎂原子分別傾向于摻雜在五配位鋁Al5c、三配位鋁Al3c的位置。相應地,在其最易摻雜位置的摻雜結合能分別為214.50、893.98、151.23 以及643.25 kJ/mol,同時,摻雜后的催化劑構型如圖3所示,其中,當鈣、鈣鋅、鈣鑭、鈣鎂在氧化鋁表面摻雜的時候,五配位鋁Al5c最易于被摻雜,與楊濤等[26]的研究結果一致。

表2 催化劑表面在不同情況下的摻雜結合能Table 2 Dopant incorporation energy of catalysts surface under different conditions

圖3 摻雜改性后催化劑表面結構俯視圖(紅色為氧原子,粉色為鋁原子,綠色為鈣原子,黃色為鎂原子,黑色為鋅原子,藍色為鑭原子)Fig.3 Top view of doped catalyst structure surfaces(O atoms are red,Al atoms are pink,Ca atoms are green,Mg atoms are yellow,Zn atoms are black and La atoms are blue)
2.2 反應物在Ca-M催化劑上的吸附
在Ca-Al-M(M= Zn, Mg, La)三元復合物催化劑催化酯交換反應過程中,由于γ-Al2O3主要起催化載體的作用,為使鋅、鎂、鑭等摻雜改性劑對鈣基活性位的調節作用更加明顯,在本節將對酯交換反應物在Ca-M 二元復合物催化劑上的吸附進行討論。在酯交換生產生物柴油的過程中,甲醇和乙酸甲酯首先吸附在催化劑表面,其中甲醇在催化劑的作用下發生解離,生成甲氧基和氫離子,甲氧基進一步攻擊乙酸甲酯上的碳氧雙鍵,生成脂肪酸烷基類酯,即生物柴油[27]。本節將主要討論甲醇和乙酸甲酯在催化劑表面吸附過程中,催化劑對甲醇上氫氧鍵及乙酸甲酯上碳氧雙鍵的影響。
分別構建甲醇在鈣基催化劑以及鈣鋅、鈣鑭、鈣鎂二元復合物催化劑上吸附的模型,構型優化如圖4 所示。甲醇在催化劑表面吸附時,氫氧鍵發生解離,吸附后甲醇的H—O 鍵長度、催化劑表面H—M 鍵長度以及吸附能如表3 所示。吸附能可以用來衡量吸附體系的穩定性,其值越大表明吸附過程中放出熱量越多,吸附越穩定。根據表3 可以看出,鋅、鎂摻雜改性劑能顯著提高甲醇在催化劑表面的吸附能,使得反應釋放更多能量,且H—O 鍵長度較吸附前(表1)明顯增加,同時H—M 鍵長度明顯減小,說明解離產生的氫原子與催化劑表面的氧原子形成新的化學鍵,最終強化了甲醇的吸附效果及活性程度;采用鑭作為摻雜改性劑時,吸附能有略微減少,但H—O 鍵長度較吸附前明顯增加,且H—M鍵長度明顯減小,也在一定程度上增加了甲醇的活化程度。
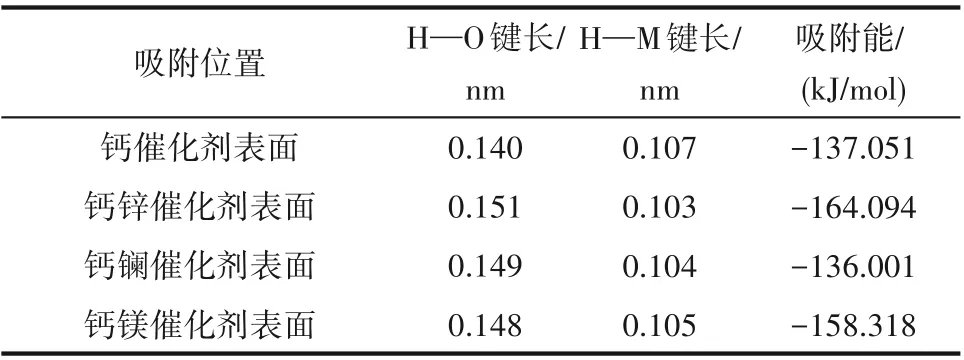
表3 甲醇吸附在催化劑表面后的鍵長和吸附能Table 3 Bond length and adsorption energy of methanol absorbed on catalyst surfaces

圖4 甲醇分子在Ca-M催化劑表面上的吸附構型Fig.4 Structure of methanol molecule absorbed on Ca-M catalyst surfaces
分別構建乙酸甲酯在鈣基催化劑以及鈣鋅、鈣鑭、鈣鎂二元復合物催化劑上的吸附模型并進行構型優化。吸附后,乙酸甲酯碳氧雙鍵C O 的鍵長變化及吸附能見表4。與催化劑對甲醇吸附結構的影響相比,催化劑對乙酸甲酯的碳氧雙鍵(表1)的作用微乎其微。當采用鋅、鑭作為摻雜改性劑時,吸附能明顯增大,有利于催化酯交換反應的進行;當采用鎂作為摻雜改性劑時,吸附能反而減小,表明反應發生的難度增加,鎂的添加不利于酯交換反應的進行。

表4 乙酸甲酯吸附在Ca-M催化劑表面后的鍵長和吸附能Table 4 Bond length and adsorption energy of methyl acetate absorbed on Ca-M catalyst surfaces
由表3、表4 可知,反應物在單一的鈣基催化劑表面以及在鈣鋅、鈣鑭、鈣鎂的二元復合物催化劑表面的吸附能分別為-257.691、-299.196、-285.695、-259.119 kJ/mol,即采用鋅、鑭、鎂作為摻雜改性劑均能增大反應物的吸附能,且活化甲醇的能力均得到增強,有利于催化酯交換反應的進行。付希[28]指出,采用溶膠-凝膠法制備的CaO-ZnO 固體堿催化劑具有較大的孔隙,能在保證催化效果的前提下具有優異的穩定性;Yan 等[29]通過多種制備方法研究出的鈣鑭二元復合催化劑擁有更高的BET 比表面積,濃度更高的強堿位點,在酯交換反應中擁有極高的催化活性;王廣欣等[30]的研究表明CaO/MgO 催化劑沉降性好,抗中毒能力強,浸出率低,相較氧化鈣催化性能得到較大改善;與計算結果相符。
2.3 甲醇在Ca-Al-M催化劑上的吸附
2.3.1 甲醇在Ca-Al-M 催化劑上的吸附能及吸附構型 根據圖3 可知,由于γ-Al2O3(110)有較高的比表面積,孔隙較大,當采用多種原子對其摻雜后,仍然保持著優異的物理結構,有利于增大醇油分子的吸附效率[31]。根據2.2 節甲醇、乙酸甲酯在Ca-M 二元復合物催化劑表面的吸附結果,采用鋅、鑭、鎂對鈣進行摻雜改性均能增強催化劑的催化效果,且甲醇在催化劑表面吸附時催化劑對其氫氧鍵活化效果最為顯著,為催化劑的主要作用部分。因此,本節將只考慮甲醇在Ca-Al-M 三元復合物催化劑表面的吸附,吸附構型如圖5 所示,吸附后甲醇中H—O鍵長度和吸附能如表5所示。
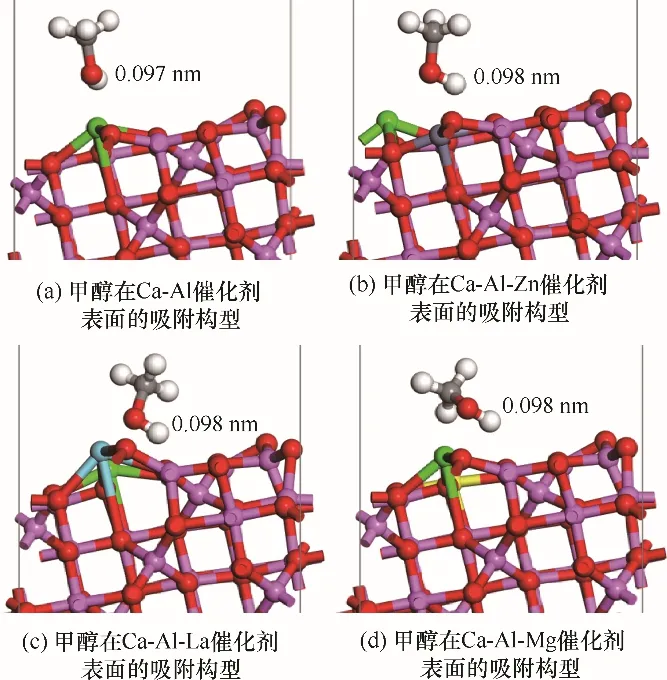
圖5 甲醇分子在Ca-Al-M 催化劑表面的吸附Fig.5 Structure of methanol molecule absorbed on Ca-Al-M catalyst surfaces
根據表5 可知,在采用鋅、鑭、鎂對鈣鋁基催化劑摻雜改性后,甲醇在催化劑表面的活化效果均得到增強,且吸附能均增大,在γ-Al2O3載體作用下,鑭摻雜改性時甲醇吸附能略微減小的缺點已經克服,催化劑催化效果變強。Norhasyimah 等[32]指出,具有三倍氧化鋁涂層的單載體氧化鈣催化劑在添加鋅物質的情況下生物柴油的轉化率顯著提高;Syamsuddin 等[33]在鈣鋁比為6∶1 的鈣鋁二元復合催化劑中添加鑭至鈣鑭鋁比為6∶2∶1 后,FAME 收率由80%上升到了96.91%;Gao 等[34]的實驗結果表明,在KF 的作用下,鈣鋁二元復合催化劑催化酯交換反應1 h 后FAME 收率可達96.6%,而鈣鎂鋁三元復合催化劑作用10 min 后FAME 收率便可達98%,與本文分析規律一致。
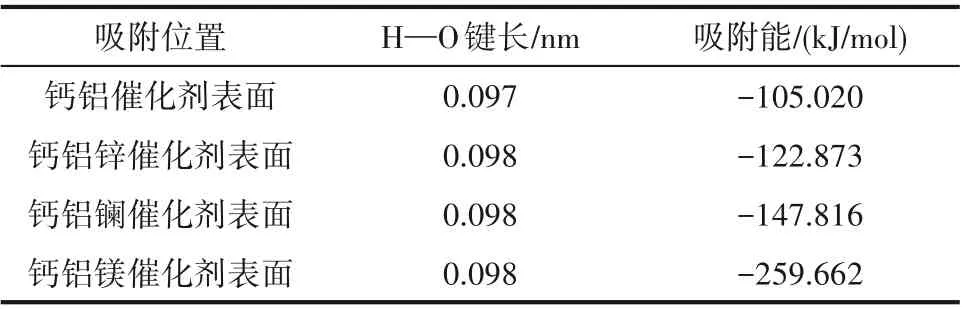
表5 甲醇吸附在Ca-Al-M 催化劑表面后的鍵長和吸附能Table 5 Bond length and adsorption energy of methanol absorbed on Ca-Al-M catalyst surfaces
2.3.2 甲醇在Ca-Al-M 催化劑上吸附的Mulliken電荷密度分析 Mulliken 電荷布局分析[35]反映吸附體系中原子與鍵在吸附前后的電子分布變化。如表6所示,吸附前后甲醇上羥基中氫原子電正性和氧原子電負性均發生變化,說明在催化劑的作用下,甲醇的電荷進行了重新分配。采用鋅作為調節劑時,氫原子電正性和氧原子電負性分別增大了0.052、0.022 e,說明甲醇得到了更好的活化;采用鑭作為調節劑時,氫原子電正性和氧原子電負性分別增大了0.078、0.034 e,也說明甲醇得到了更好的活化;采用鎂作為調節劑時,氫原子的電正性增大了0.026 e,氧原子的電負性減小了0.005 e,但增大數值明顯大于減小數值,仍說明甲醇得到了更好的活化。將甲醇上各個原子電荷密度相加得到甲醇分子總電荷密度的變化值,說明甲醇在催化劑表面吸附之后失去電子,且相較于鈣鋁Ca-Al二元復合物催化劑,使用鋅、鑭、鎂摻雜改性后,Ca-Al-M 三元復合物催化劑失去電子的數量增加,采用鋅、鑭、鎂進行調節后,甲醇失電子數分別增加0.016 e、0.044 e、0.006 e。以上分析表明,甲醇分子吸附到催化劑表面時,甲醇與催化劑表面活性中心之間存在電子傳遞過程,從而增強了甲醇分子的吸附穩定性,同時也活化了甲醇分子,進一步摻雜改性后,甲醇與催化劑表面的電子轉移數增加,說明甲醇的活化程度增強[36]。結果表明,采用鋅、鑭、鎂作為調節劑時,更有利于吸附反應的發生,甲醇的活化得到增強,這與吸附能和結構分析的結果一致。
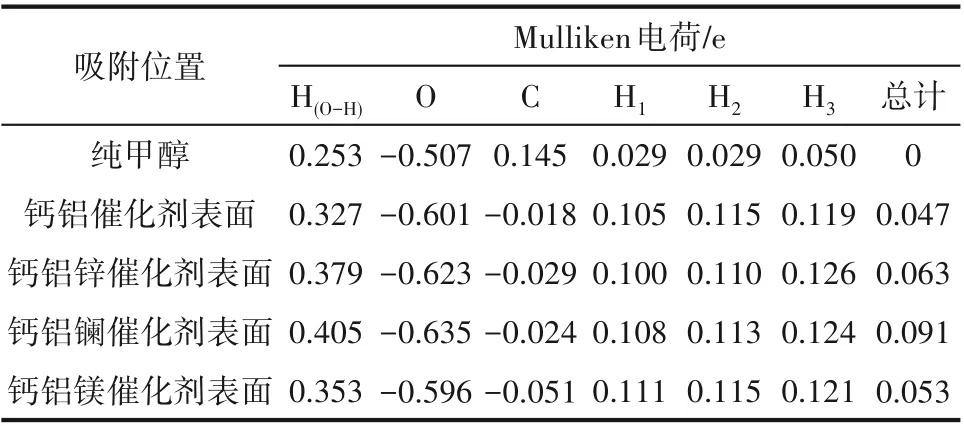
表6 甲醇吸附在Ca-Al-M催化劑表面的Mulliken電荷布局Table 6 Mulliken atomic charge populations for methanol adsorption on Ca-Al-M catalyst surfaces
3 結 論
(1)分別優化了鈣鋁、鈣鋁鋅、鈣鋁鑭、鈣鋁鎂復合催化劑的幾何構型,結果表明鈣鋁二元復合催化劑中鈣原子傾向于摻雜在五配位鋁的位置,鈣鋁鋅三元復合催化劑中鈣原子、鋅原子分別傾向于摻雜在四配位鋁、五配位鋁的位置,鈣鋁鑭三元復合催化劑中鈣原子、鑭原子分別傾向于摻雜在三配位鋁、五配位鋁的位置,鈣鋁鎂三元復合催化劑中鈣原子、鎂原子分別傾向于摻雜在五配位鋁、三配位鋁的位置。
(2)鋅、鑭、鎂對鈣進行摻雜改性能有效地增強其對甲醇的活化能力,當反應物在催化劑表面吸附時,吸附能均增大,其中甲醇在催化劑表面的吸附是酯交換反應的決速步驟。
(3)γ-Al2O3憑借其優異的結構特征,作為催化反應的載體能有效增強催化效果。甲醇在鈣鋁二元復合催化劑上吸附時,采用鋅、鑭、鎂進行摻雜改性后,甲醇在催化劑表面的吸附能明顯增大,甲醇活化程度增強。通過對甲醇進行Mulliken電荷密度分析,進一步驗證了鋅、鑭、鎂增強催化劑催化能力的效果。
符 號 說 明
EAl——Al單原子能量,kJ/mol
EAl2O3——氧化鋁表面能,kJ/mol
Eads——吸附能,kJ/mol
Edope——摻雜結合能,kJ/mol
EM——M單原子能量,kJ/mol
EMAl2O3——M原子摻雜Al2O3表面后的總能量,kJ/mol
EMNAl2O3——M 和N 原子摻雜Al2O3表面后的總能量,kJ/mol
Emolecule——分子吸附前的總能量,kJ/mol
EN——N單原子能量,kJ/mol
Esurface——催化劑表面能,kJ/mol
Etotal——催化劑表面吸附分子后的總能量,kJ/mol
猜你喜歡
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
中國塑料(2016年12期)2016-06-15 20:30:07
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
中國塑料(2016年5期)2016-04-16 05:25:36
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
