經線粒體途徑相關信號通路對成骨細胞的影響
2020-09-07 01:13:24張騰魏鶴翔張永吳定張成俊
中國骨質疏松雜志 2020年8期
張騰 魏鶴翔 張永 吳定 張成俊*
1.蘭州大學第二臨床醫學院,甘肅 蘭州 730000
2.蘭州大學第二醫院,甘肅 蘭州 730000
骨質疏松(osteoporosis,OP)是以骨微觀結構改變、骨脆性增加、骨量減少為特征的全身代謝性骨質病變。有關研究表明,老年人及絕經后的婦女是骨質疏松癥的高發人群。目前全球多數國家已開始步入人口老齡化,因此研究有關骨質疏松的微觀機制及防治藥物具有重大的臨床意義。
骨質疏松主要受成骨細胞(osteoblast,OB)的凋亡或破骨細胞(osteoclast,OC)的增殖影響發生。導致成骨細胞凋亡的途徑中,線粒體凋亡途徑是導致成骨細胞凋亡的主要途徑。通過線粒體途徑及相關通路,可進一步探明骨質疏松的發病機制,便于更好的治療骨質疏松。
1 線粒體凋亡途徑
線粒體是為細胞供能的主要細胞器,為細胞正常活動供應能量、傳遞信號,參與細胞凋亡等過程。線粒體通透性轉位孔(permeability transition pore,PT孔)的開放是引發線粒體凋亡途徑的主要原因,PT孔是由線粒體外膜上的陰離子通道(voltage dependent anion channel,VDAC)和線粒體內膜上的腺苷酸轉位因子(adenine nucleotide translocator,ANT)等組成的。PT孔開放后,線粒體內外膜通透性改變,電、化學梯度不能維持,跨膜電位ΔΨm下降,水分進入內膜,導致線粒體基質腫脹,釋放出膜間隙的細胞色素C(Cytc)、Smac蛋白(the second mitochondrial derived activator of caspase)和凋亡誘導因子(apoptosis induce factor,AIF)[1-2]。
線粒體釋放出細胞色素C后,細胞色素C與凋亡蛋白酶活化因子(apoptosis protease activating factor 1,ApaF-1)結合,位于細胞質中的ApaF-1再與ATP結合改變構象,招募caspase 9前體,caspase 9前體自我剪切活化為有活性的caspase 9蛋白,有活性的caspase 9進一步激活caspase 3,啟動caspase級聯反應。caspase蛋白可激活CAD(caspase- activated Dnase),CAD是核酸酶的一種,可引起細胞骨架蛋白和核酸水解,致使細胞凋亡[2]。
從線粒體釋放出來的凋亡誘導因子AIF,與細胞色素C和ApaF-1通過caspase級聯反應誘導凋亡有所不同,可直接進入細胞核作用于核酸,在相關酶催化下引起DNA大規模斷裂和染色質濃縮[4]。AIF可不用caspase蛋白引起凋亡[3]。
Smac蛋白位于線粒體膜間隙中,具有調控凋亡的功能。在線粒體收到凋亡信號時,Smac蛋白進入胞質與凋亡抑制蛋白結合,阻礙其與caspase家族蛋白的結合,從而起到促進凋亡的作用[5,41]。Smac蛋白不能主動引發凋亡,但能促進凋亡。
在通過線粒體途徑凋亡的過程中,有一組BcL-2家族蛋白可以參與調控,按功能可分為促凋亡蛋白和抗凋亡蛋白,促凋亡蛋白主要有Bax、Bak、Bid、Bik等,抗凋亡蛋白有BcL-2、Bcl-xl、Bcl-w等[7,42]。促凋亡蛋白主要通過增加VDAC通透性,抗凋亡蛋白通過與促凋亡蛋白競爭結合相關位點或阻止促凋亡蛋白與相關位點結合來發揮作用[6]。
2 線粒體途徑相關信號通路
2.1 PI3K/Akt信號通路
PI3K(phosphatidylinositide 3-kinases),是一種由調節亞基 p85 和催化亞基 p110 構成的二聚體蛋白質。Akt又稱蛋白激酶PKB(protein kinase B)是一種絲氨酸/蘇氨酸特異性蛋白激酶,擁有PH結構域。PI3K通路受細胞內外信號調節,當細胞因子等信號刺激受體酪氨酸激酶(RTK)和G蛋白偶聯受體(GPCRs)時,胞質區酪氨酸殘基活化,p85亞基轉移至質膜內表面,磷脂酰肌醇 4,5-二磷酸 (phosphatidylinositol 4,5-diphosphate,PIP2)受p110磷酸化變為磷脂酰肌醇 3,4,5-三磷酸(phosphatidylinositol3,4,5 -trisphosphate,PIP3)[8],PIP3募集擁有PH結構域的Akt至質膜內面,Akt的Thr308殘基被3-磷酸肌醇依賴性蛋白激酶 1(PDK1)磷酸化,Ser473殘基被哺乳動物雷帕霉素目標復合物(mTORC2)磷酸化,使得Akt被完全激活[9-10]。完全激活后的Akt可作用于GSK-3β、BcL-2家族蛋白、FoxO等的相關基因,調節細胞增殖、凋亡、遷移、生長代謝[19,43]。PI3K信號通路的激活,促進了成骨細胞分化標志物堿性磷酸酶ALP、BMP-2的表達,有利于成骨細胞的增殖分化[20]。Xue等[40]研究發現,紅景天甙可以作用于地塞米松誘導的成骨細胞,可激活PI3K/Akt通路,降低了Bax、細胞色素C、cleaved caspase-3,、cleaved caspase-9的表達,同時提高了BcL-2和p-Akt的表達。
有研究[16]表明,在流體剪切力抑制TNF-α誘導的凋亡實驗中,激活PI3K-Akt通路后,Akt活化后降低了caspase-3和Bim的表達,抑制了成骨細胞的凋亡。PI3K-Akt通路通過調控caspase、BcL-2家族蛋白的方法抑制凋亡。除此之外,PI3K還可通過磷酸化GSK-3β,促進β-catenin的堆積,進而增強經典Wnt信號通路[21],促進細胞增殖。
PI3K通路對成骨細胞的作用主要表現在促進增殖分化和抑制凋亡兩方面,在骨組織層面表現為促進骨形成,增加骨量的作用。該通路對治療骨質疏松方面具有重大意義,而Akt是該通路的核心靶點,故未來可研究作用于Akt的藥物來治療骨質疏松。
2.2 MAPK信號通路
絲裂原活化蛋白激酶(MAPK)是一族高度保守的絲/蘇氨酸蛋白激酶家族,在細胞的許多生命活動都起了重要作用。MAPK信號傳導由MAPKKK、MAPKK、MAPK的三個核心激酶逐層級聯化反應組成。其中MAPK信號通路主要有四種,以其組件命名為JNK通路、ERK5通路、p38通路、ERK1 /2通路[11]。
2.2.1JNK通路:Jun氨基末端激酶((c-Jun N-terminal kinase,JNK)是JNK通路的核心物質,關鍵靶點。JNK的編碼基因包括三種:JNK1、JNK2、JNK3。JNK通路可由細胞因子、細胞毒性物質、電離輻射等激活,JNK通路激活順序一般為ASK1、MEKK1激活MKK4/7,MKK4/7又激活JNKS。JNKS又可以激活部分BcL-2家族成員以及提高早期生長反應因子(early growth response 1,EgR-1)、基環氧酶2(cyclo-oxyge-nase-2,COX-2)、骨橋素和c-fos 的表達[32,44]。Chen[22]等的研究表明,補骨脂素可抑制JNK途徑,下調p-ASK、p-JNK、Bax的表達,上調BcL-2的表達來抑制成骨細胞凋亡。Guo等[28]的研究表明,通過LPS激活JNK通路,胞內p-JNK水平提高,提升了Bad和caspase-3的表達,降低了BcL-2的表達,激活caspase-3,誘使成骨細胞自線粒體途徑凋亡。JNK不僅是線粒體途徑誘發凋亡的關鍵物質,也可參與誘發內質網途徑導致凋亡。阻斷JNK通路可顯著抑制成骨細胞的凋亡,對治療骨質疏松有積極意義。
2.2.2p38通路:激活p38通路的物質與JNK相似,由炎性因子(TNFα,IL-1 或 IL-6)及應激刺激(紫外輻射、熱或滲透性休克)等刺激。P38通路的激活途徑一般由Mlk1-3、MEKK1-4、TAK、ASK1 /2激活MEK3和 MEK6,再由MEK3和 MEK6進一步激活P38。P38 激活后可激活轉錄因子NF-κB、ATF-2、Elk-1、MEF-2、Stat1、Max、p53 等[23]。Yu等[24]研究表明,地塞米松誘導成骨細胞凋亡的過程中,激活了p38信號通路,用葛根素抑制p38通路,降低p-p38MAPK的表達,可見caspase-3表達下調。此外,彭鎏[35]研究發現,激活的p38 MAPK可引起IκB激酶增加,激活NF-κB信號通路,促進細胞凋亡。Bai等[38]研究表明,在體外用大劑量地塞米松處理成骨細胞,可引起ASK1和p38表達水平升高,激活p38通路,促使成骨細胞凋亡。由此可見,p38通路可以促使成骨細胞凋亡,抑制骨形成,促進骨質疏松的發展進程。研發抑制p38通路的藥物有利于預防或治療骨質疏松。
2.2.3ERK1/2通路:ERK1/2通路受細胞因子、生長因子、激素、滲透壓等刺激激活,激活途徑是先激活Ras再激活Raf,Raf通過MEK1/2將信號傳遞給ERK1/2并激活ERK[11]。ERK使許多與胞質胞膜相連的底物c-RaF-1、MEK和磷脂酶A2等磷酸化,且移入胞核可磷酸化一系列促使成骨細胞增殖的轉錄因子如ELK-1、SAP、AP-1等,促使細胞增殖[33]。HK等[25]研究表明,膠原蛋白水解物可誘使ERK1/2磷酸化,促進成骨細胞的分化增殖。給隨后建立了骨質疏松模型的小鼠口服膠原蛋白水解物,小鼠腰椎骨密度上升。Li 等[34]的研究表明,激活ERK1/2后,BcL-2表達下調,Bax表達上調,Bax/BcL-2比值升高,促使細胞凋亡。因此,這一通路對成骨細胞的具體作用存在爭議,尚不明確。
2.2.4ERK5通路:ERK5信號通路可由高滲透壓、生長因子、流體剪切力、氧化劑等激活。ERK5的激活途徑為激活MEKK2/3后,在通過MEK5激活ERK5。Bin等[27]研究表明,流體剪切力通過ERK5途徑抑制TNF-α誘導的成骨細胞凋亡,活化的ERK進入胞核導致Bad磷酸化,p-Bad被胞質中的蛋白阻隔,進入不了線粒體。通過減少線粒體中的Bad,可以抑制其引起的線粒體途徑caspase-3的活化。此外p-ERK5還可激活 p-Akt,減少Bim 的表達,對抗TNF-α導致的凋亡[26]。有關研究往往通過ERK5通路對抗TNF-α等誘導的凋亡。見圖1。
2.3 Wnt/β-catenin信號通路
信號通路未被激活時,β-catenin被糖原合酶激酶(glycogen synthase kinase -3β,GSK-3β)、Axin、蛋白激酶 1(casein ki-nase 1,CK1)、腺瘤病大腸桿菌(APC)、蛋白磷酸酶2 A(PP2A)等組成的蛋白復合體結合,使β-catenin被定向泛素化,從而被降解[13]。Wnt信號被激活時,與Frizzled和低密度脂蛋白相關蛋白(LRP5/6)等結合,蛋白復合體被磷酸化,招募Axin從而使GSK-3β與蛋白受體復合物分離[14]。β-catenin不能被失去了結合物的GSK-3β磷酸化,β-catenin堆積在細胞中,進入細胞核后與LEF/TCF轉錄因子家族結合,啟動下游靶基因 (如c-myc、Cyclin D1等) 的轉錄[12,45]。Kang[36]的研究表明,激活后的Wnt/β-catenin信號通路,使BcL-2轉錄上調,抑制caspase-8及caspase-3活性,抑制細胞凋亡。Wei等[29]在研究敲除了β-catenin基因的小鼠時發現,激活β-catenin可抑制破骨細胞的分化。另有研究[30]表明,可通過Wnt/β-catenin信號通路調節脂肪來源的干細胞促使其向成骨細胞分化。Man等[39]研究發現,熊果苷可提高β-catenin和Runx2的表達水平,激活經典Wnt信號通路,促使成骨細胞增殖分化。
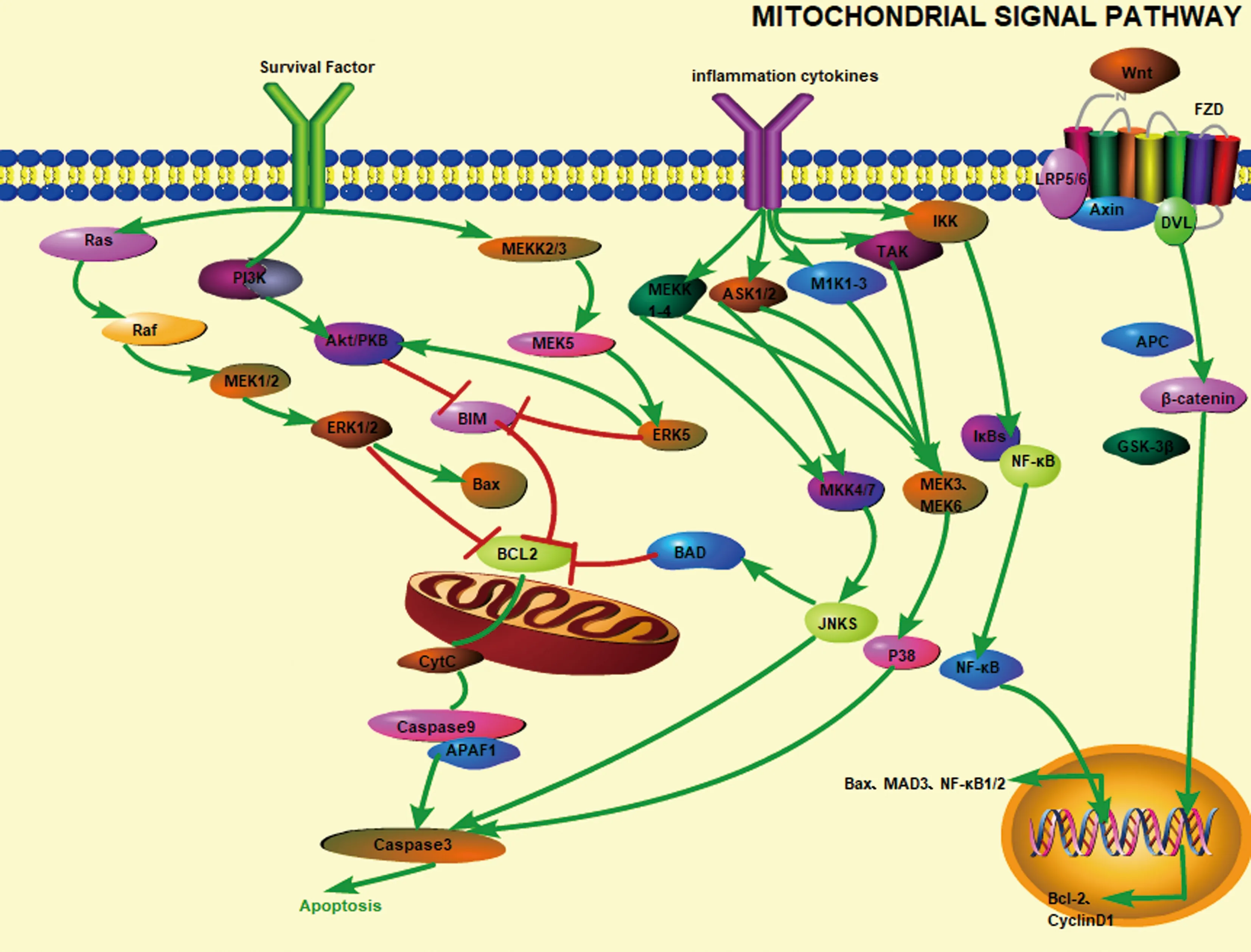
圖1 成骨細胞內各相關信號通路、細胞因子經線粒體途徑調控細胞凋亡示意圖Fig.1 Schematic diagram of regulation of apoptosis by mitochondrial pathway in various related signaling pathways and cytokines in osteoblasts
激活經典Wnt信號通路的主要作用是促進成骨細胞增殖分化[31],抑制破骨細胞分化,且該通路促使成骨細胞增殖的作用較強,是影響骨量增減的關鍵通路。而影響該通路的開關因素,就是β-catenin的堆積程度。從這個角度研發針對調節β-catenin的藥物可以較好的調節骨量變化。
2.4 NF-κB信號通路
NF-κB是一個核轉錄因子家族,P50、P52、REL、REL-A和REL-B組成了該家族。該家族成員除REL-B外,均可相互結合形成同二聚體或異二具體。REL-B只可以與p50或p52組成二聚體。通路未被激活時NF-κB通常與IκB結合形成復合物,使NF-κB失去活性。在細胞因子類(TNF-α、IL-1、IL-2),病度感染及X射線及紫外射線等對細胞施加刺激后,以上刺激激發的第二信使促使IκB降解,NF-κB產生活性。激活的NF-κB轉移向核中,選擇上調NF-κB1(編碼plo5)、NF-κB2(編碼pl00)、p Rel、BcL-3 和 MAD3(編碼IκBα)等基因[18,46]。彭鎏[35]的研究表明,激活的NF-κB轉入核內與Bax基因上的位點結合,促進Bax的表達,從而Bax/BcL-2比值上升,促使細胞凋亡。龐同濤[37]的研究也表明,NF-κB表達增加、通路激活,可使下游的BcL-2表達下調,促進成骨細胞凋亡,抑制增殖。
經典NF-κB通路的激活主要降低成骨細胞的代謝能力,還可通過阻礙Smad蛋白發揮作用來降低成骨細胞的增殖分化能力,促進成骨細胞凋亡為其次要作用。
3 小結與展望
綜上,本文介紹了線粒體途徑及相關信號通路對成骨細胞的影響。其中,PI3K/Akt、ERK5、Wnt/β-catenin信號通路促進成骨細胞增殖分化,JNK通路、P38、NF-κB通路抑制成骨細胞增殖分化。大多信號通路可直接調節線粒體途徑中的caspase、BcL-2家族蛋白等關鍵靶點的調節,從而通過抑制凋亡來促使凋亡,還可通過調控細胞增殖分化過程中的關鍵物質及其轉錄因子來影響成骨細胞。
當前有關成骨細胞的研究,仍將細胞的增殖、凋亡等活動籠統地劃分在三大途徑里。未來有關成骨細胞的研究應更多側重于各途徑信號通路之間的聯系影響上,力求將影響成骨細胞增殖、凋亡的各物質包容在一個統一的體系中。在相關疾病的治療上,深入研究不同藥物、激素、胞內外因子作用于各通路關鍵靶點上的效果,將已有的科研成果轉化為臨床成果,以解決骨質疏松等病癥的治療問題。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
科技傳播(2019年22期)2020-01-14 03:06:54
中學生數理化·七年級數學人教版(2019年10期)2019-11-25 07:33:58
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
中學生數理化·高一版(2018年9期)2018-10-09 06:46:50
電子制作(2018年11期)2018-08-04 03:25:42
湖南教育·C版(2018年3期)2018-06-05 16:54:36
