一種高純度5-氟尿嘧啶合成工藝研究
2020-09-24 03:24:16陳小林
科技視界 2020年26期
陳小林
0 引言
5-氟尿嘧啶(5-FU)由Duschinsky 等[1]于1957 年首次合成,是一個重要的抗腫瘤藥物,也是氟代嘧啶類抗腫瘤藥物的重要中間體。目前氟尿嘧啶的合成方法(根據Scifinder 調研)可歸納為3 種方法:直接氟化法,縮合環合法及其他方法。直接氟化法是以尿嘧啶為原料,與惰性氣體(N2)稀釋的氟氣或活性含氟化合物反應直接合成目標產物(5-氟尿嘧啶)[2-4]。此法工藝簡單,收率高,但因氟氣來源少,價格貴,氟氣毒性大、對設備的要求高等原因,在產業化生產過程中應用較少。其他方法主要是以含氮雜環化合物的衍生物為起始原料,此法原料難得,成本高,不適宜產業化[5-6]。
目前主流工業化路線為縮合環合法,以氟乙酸甲酯為起始物料,經與甲酸乙酯在甲醇鈉的催化下等到氟代丙醛酸甲酯烯醇式鈉鹽,此中間體與甲異脲(固體或者液體)環合成2-甲氧基-5-氟尿嘧啶,后經酸水解、重結晶等過程得到5-氟尿嘧啶[7-10]。此法路線長,效率低,但原料廉價(國內有大宗商品),反應條件溫和(未見嚴苛的反應條件),易于產業化,總體成本低廉而得到國內生產廠家的青睞。
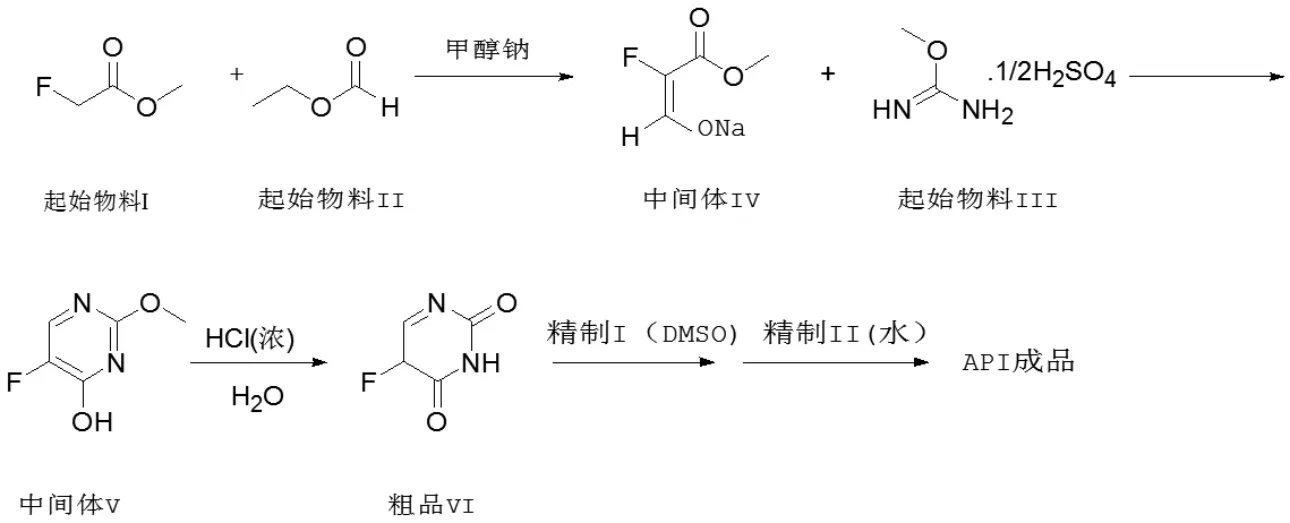
圖1 高純度5-氟尿嘧啶合成工藝反應路線示意圖
作者以外購氟乙酸甲酯為起始物料,在甲醇鈉的催化下與甲酸乙酯縮合得到烯醇式鈉鹽(中間體IV),不純化直接與固體甲異脲硫酸鹽環合成中間體V,然后在稀鹽酸下水解得到粗品,粗品經過二甲亞砜、水2 步精制,得到HPLC 純度>99.9%的高純度成品。反應路線如圖1 所示。
1 試驗
1.1 試劑與儀器
上述合成路線中用到的試劑及儀器如下:
1.1.1 起始物料
1.1.2 其他物料
濃鹽酸(HCl)-上海國藥-AR。
1.1.3 溶劑
無水甲醇(CH3OH)-上海國藥-AR。
純化水(H2O)-自制-中國藥典2015 年版。
1.1.4 輔助材料
藥用活性炭C-上海長興活性炭有限公司-藥用級。
1.1.5 主要儀器及型號
天平(Sartorious-型號:BSA4202S-CW)。
真空烘箱(上海禾氣玻璃儀器有限公司:DZF-6020)。
帶加熱磁力攪拌器(上海禾氣玻璃儀器有限公司:DF-101S)。
隔膜真空泵(上海禾氣玻璃儀器有限公司:M22CHT)。
高壓液相儀(water-型號:e2695-2998PDA)。
液質聯用儀(Q-Tofmicro-型號:Q-Tofmicro)。
核磁共振波譜儀(BRUKER-型號:BRUKER-400)
1.2 合成方法
1.2.1 氟代丙醛酸甲酯烯醇式鈉鹽(化合物IV)的合成
(1)投料物料配比。
(2)合成步驟。
往干燥的三口反應瓶(3L)內加入甲苯,開始攪拌,氮氣置換三次;繼續往反應瓶內加入固體甲醇鈉,然后內溫降至0~15℃;往反應瓶內繼續滴加甲酸乙酯液體,控制內溫0~20℃,1 h 內滴加完畢;滴加氟乙酸甲酯液體,控制內溫不超過30℃,1 h 內滴完;繼續攪拌升溫至30~40℃,保溫反應9 h。此反應液無須處理直接進行下一步。

表1 化合物IV 投料物料配比表
1.2.2 2-甲氧基-5-氟尿嘧啶(中間體V)的合成
(1)投料物料配比。

表2 中間體V 投料物料配比表
(2)合成步驟。
上述反應液轉入另一個5 L 的三口瓶中,氮氣置換三次,將瓶內溫度冷卻至10℃,開啟攪拌,依次加入甲醇鈉溶液、固體甲異脲,控制內溫35~45℃。保溫攪拌9 h 后,取樣送中控,檢測含量,若反應液中產物含量≥10.0%,則終止反應,進行下一步操作。若<10.0%,則繼續反應,并每隔3 小時取樣直至含量≥10.0%。
后處理:加入水1.6 L,攪拌30 min 使反應液溶清,停止攪拌,靜置分層,將下層水相轉入至另外一個反應瓶中內。水相冷卻至20~35℃,滴加濃鹽酸,調節pH 至3~4,析出大量淡黃色固體,降溫至10℃,繼續攪拌30min 后過濾。濾餅折干后約190.5 g,摩爾收率:60.6%。MS ESI+=145.10(M+1);1H-NMR(400 MHZ,DMSO)δ∶3.863(s,3H),7.852-7.861(d,J=3.6,1H),12.853(s,1H)。
1.2.3 粗品(VI)的合成(1)投料物料配比。
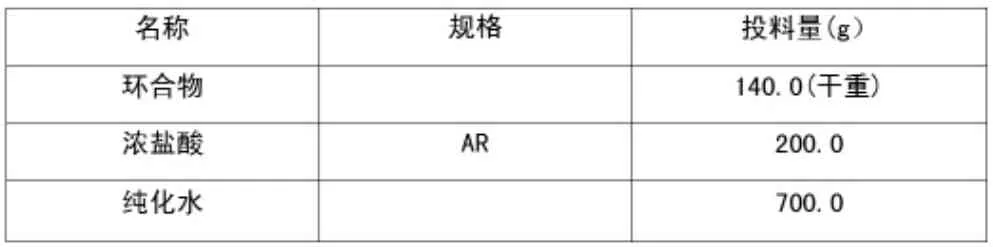
表3 粗品(VI)投料物料配比表
(2)合成步驟。
往三口瓶(3 L)中加入純化水(200 ml),攪拌下加入2-甲氧基-5-氟尿嘧啶化合物;氮氣置換3 次后開始滴加鹽酸溶液(4M)),1 h 左右滴完;內溫升至50~60℃,保溫反應3 h;取樣送檢(HPLC 中控),當液相結果中環合物含量≤5%(面積歸一法)時,反應結束后,內溫冷卻至5~15℃,過濾,得粗品,粗品折干重月116.2 g,摩爾收率:91.9%。
1.2.4 粗品精制
1)投料物料配比。
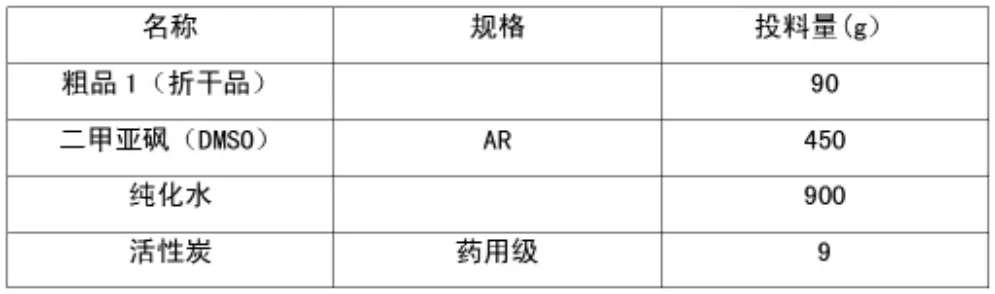
表4 粗品精致投料物料配比表
2)合成步驟。
(1)二甲亞砜精制。
將DMSO 和粗品加入單口反應瓶內,攪拌升溫至內溫70~80℃,反應液溶清,保溫攪拌1h;開始降溫至內溫降至20~25℃,保溫攪拌3h 后過濾,得濾餅,濾餅無須干燥直接投下一步。
(2)水精制
將二甲亞砜精制得到的粗品加入純化水中,攪拌升溫至回流,反應液溶清,加入活性炭;回流保溫30mins,趁熱過濾,濾液緩慢降溫至T=10℃,析晶2 小時后過濾,濾餅50℃真空干燥16 h后,得成品64.3 g,摩爾總收率:71.4%。m.p.284.1-284.4(藥典:282-286℃)。MS ESI+=131.10 (M+1),153(M+Na);1H-NMR(400MHZ,DMSO)δ∶7.721-7.736(d,J=6.0,1H),10.702(s,1H),11.489(s,1H)。
2 結果與討論
2.1 2-甲氧基-5-氟尿嘧啶(中間體V)的條件優化
2.1.1 縮合反應
由于化合物IV 是烯醇式鈉鹽,目前沒有很好的分析方法來監控,故這步優化后直接做成中間體V,優化指標主要是通過中間體V 的收率及純度來衡量。主要優化參數為:
堿的種類(甲醇鈉、氫氧化鈉、三乙胺)、堿當量(1.5eq、2.0eq、2.2eq、2.5eq)、縮合反應溫度(25±5℃、35±5℃、50±5℃)、甲酸乙酯當量(2eq、2.25eq、3.0eq)、縮合反應時間(4 h、9 h、16 h),確定這步最佳反應條件為:甲醇鈉(2.2eq)、反應溫度(35±5℃)、甲酸乙酯用量(2.25eq)、反應時間(9 h)。
2.1.2 環合反應
環合反應可以通過高壓液相來監控,通過生成中間體V 的含量及純度兩個指標來優化各參數。具體優化參數如下:
環合反應溫度(25±5℃、40±5℃、50±5℃)、環合反應甲醇鈉當量(0.7eq、1eq、1.3eq)、固體甲異尿當量(1eq、2eq、3eq),確定最優工藝參數為:環合溫度(40±5℃)、環合堿當量(1.3eq)、固體甲異尿當量(2eq)。
此兩步摩爾總收率:60.6%,中間體V 的HPLC 純度為95.0%以上。
2.2 粗品(VI)的條件優化
此步水解反應主要考察不同的酸水解(濃鹽酸、硫酸、乙酸),溶劑體積(10 V、5 V、3 V),反應溫度(35±5℃、45±5℃、55±5℃)等工藝參數。
確定最佳工藝參數為:水解酸用濃鹽酸,水體積為5 V,反應溫度為55±5℃。
這步反應摩爾收率:91.9%。
2.3 精制的條件優化
第一次精制溶劑篩選(DMF、DMSO、H2O、丙酮),溶劑體積篩選(3 V、5 V、10 V);第二步精制溶劑主要篩選溶劑體積(5 V、10 V、15 V),藥用活性炭用量(5%、10%、15%)。
確定最佳工藝參數:第一步精制溶劑選5 V 的二甲亞砜重結晶;第二步精制溶劑選10 V 的水,活性炭選10%的量。兩步重結晶摩爾收率為:71.4%,HPLC 純度>99.9%。
3 總結
通過對本路線各中間體步驟的優化,總路線的摩爾收率39.8%,HPLC 純度99.9%以上(面積歸一法),整條反應路線未使用到特殊反應條件及試劑,適合工業化,且通過此路線得到的產品質量超中國藥典及歐洲藥典標準。
