成人面部叢狀血管瘤一例
2020-09-25 05:58:02馮曦微
中國麻風皮膚病雜志 2020年10期
馮曦微 李 凡 王 琳
四川大學華西醫院皮膚性病科,四川成都,610041
臨床資料患者,女,54歲。因右下頜角多發暗紅色斑疹伴刺痛1年余于2019年10月至我科就診。患者1年前發現右下頜角多個暗紅色斑疹,最大約2 cm×2 cm,伴刺痛,光照、溫度升高時痛感明顯,先后于外院診斷為粉刺、皰疹、盤狀紅斑狼瘡等疾病,并予以患處外涂糖皮質激素乳膏、米諾環素乳膏等治療,皮損無好轉,且范圍逐漸擴大,累及頰部。患者平素身體健康,否認高血壓、糖尿病、冠心病等基礎疾病,否認家族遺傳病史,無類似疾病家族史。系統性檢查未見異常。
體格檢查:系統檢查未見明顯異常。皮膚科檢查:右面部下頜區域散在多個暗紅色斑疹,表面光滑、邊界不清,觸之質韌,無觸痛(圖1)。實驗室檢查:血常規、生化、凝血檢查未見明顯異常。組織病理示:表皮大致正常,真皮內散在毛細血管小葉成簇狀分布,小葉內可見內皮細胞緊密排列,形成不規則管腔。內皮細胞大小一致,無異型性。部分小葉周圍可見擴張淋巴管(圖2)。免疫組化染色:血管內皮細胞CD31(+),CD34(+),D2-40,SMA灶狀陽性(圖3)。
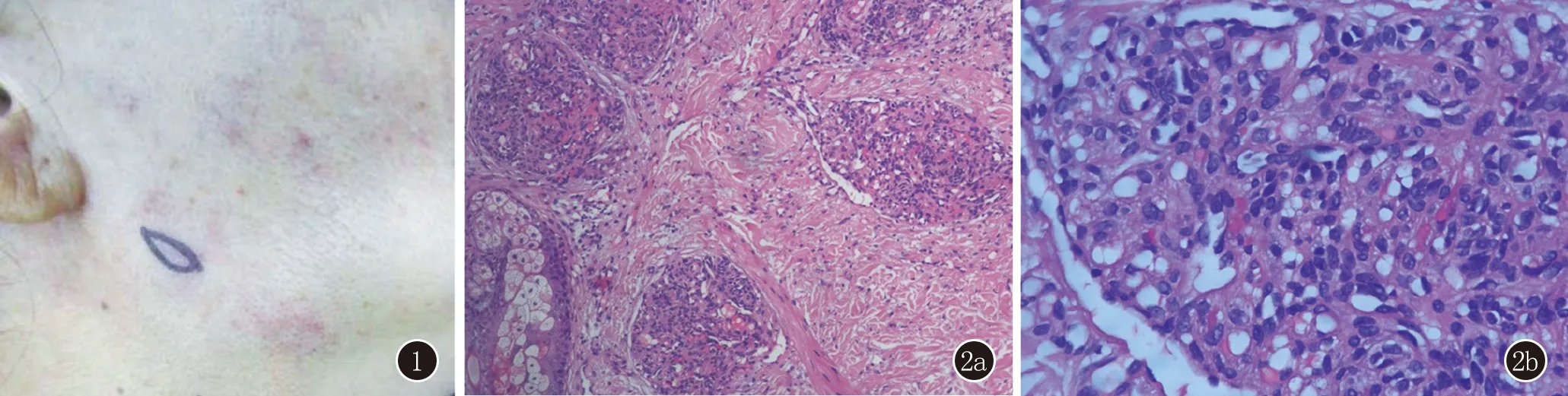
圖1 右下頜面部多發暗紅色斑疹、斑片 圖2 2a:真皮內散在成簇毛細血管小葉,小葉周圍見擴張淋巴管(HE,×100);2b:小葉內見梭形細胞緊密排列,無明顯異型性(HE,×400)
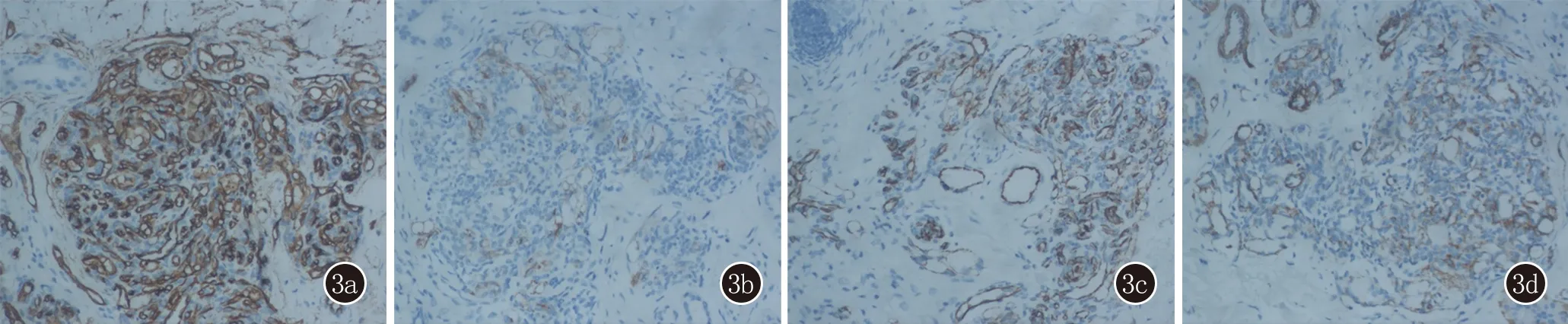
圖3 3a:CD34陽性(SP,×200); 3b:CD31陽性(SP,×200);3c:D2-40局灶陽性(SP,×200);3d:SMA局灶陽性(SP,×200)
診斷:叢狀血管瘤。患者拒絕治療,目前隨訪中。
討論叢狀血管瘤(tufted angioma,TA)是一種少見的血管良性腫瘤,1949年Nakagawa首先報道,并命名為Nakagawa血管母細胞瘤,1976年被Wilson Jones首次描述為叢狀血管瘤。
TA通常在5歲以前發病,偶見于成人,無明顯性別差異,好發于頸部、軀干上部,也有報道發生在口腔黏膜、大腿和肢端[1-3]。臨床表現常為形狀大小各異的紫色或暗紅色斑片、丘疹或斑塊,可觸及皮下結節。部分皮損處可有多毛、多汗的表現。少部分患者有疼痛感,可在日曬后加重,推測可能與血管內皮細胞、肌上皮細胞收縮有關。本病生長緩慢,最終穩定在一定大小,尚無惡變報告。部分兒童可在6個月到2年內自行消退。TA還可伴發卡梅綜合癥(Kasabach-Merritt syndrome,KMS),KMS是一種表現為血小板減少、凝血功能異常、低纖維蛋白原血癥的少見凝血病,嚴重者可危及患者生命。故診斷為TA的患者需要檢查血常規和凝血功能。本病的病因和發病機制尚不清楚,部分學者猜測與高水平雌激素刺激血管增生有關[4]。有學者將叢狀血管瘤視為一疾病譜,并根據預后的不同簡單分為無并發癥的TA、無血小板減少但伴有慢性凝血障礙的TA、伴有KMS的TA[5]。
本病確診主要靠組織病理學檢查,典型表現是由上皮樣或梭形細胞組成的毛細血管小葉呈“炮彈狀”散在浸潤真皮各層,邊界清楚,周圍常繞以纖維結締組織。血管小葉內可見含有紅細胞的狹長裂隙,外周常散布一些擴張的淋巴管。內皮細胞無異型性。本病需要與鮮紅斑痣、嬰兒血管瘤、卡波西血管內皮瘤、化膿性肉芽腫、細菌性血管瘤、小汗腺血管錯構瘤等鑒別。有研究發現D2-40在TA和卡波西血管內皮瘤的不同表達對于鑒別兩者有重要意義,在叢狀血管瘤中,D2-40僅在血管小葉周圍擴張的淋巴管中表達,在簇狀增生的毛細血管內不表達,而卡波西血管內皮瘤恰恰相反[6]。本例患者的D2-40在外周擴張的淋巴管灶性表達,而在毛細血管小葉內表達陰性,支持TA的診斷。
小病灶可用手術切除[7],冷凍療法、電子束照射和脈沖染料激光等也有一定療效[8]。系統以糖皮質激素作為一線治療手段,但不良反應多,易復發,不推薦作為首選方式[9],其他有效治療包括長春新堿、非選擇性β受體阻滯劑等。有多例報道稱口服西羅莫司療效確切,且能有效遏制KMS的發生發展[10]。但此病尚無統一的標準治療方案。由于本病呈良性進展,且有自發消退傾向,故也可選擇不予處理,定期監測血常規和凝血功能。本例為成年患者,皮損發生于面部,臨床罕見,易誤診,組織活檢后確診。患者屬無并發癥TA,拒絕治療,目前繼續隨訪中。
