5-甲氧基-2-硝基-4-(3-(吡咯烷-1-基)丙氧基)苯甲酸甲酯的合成
2020-10-26 08:51:28高艷蓉唐文強問娟娟仝紅娟
化學與生物工程 2020年10期
高艷蓉,唐文強*,問娟娟,仝紅娟,劉 斌
(1.陜西國際商貿學院醫藥學院,陜西 西安 712046;2.陜西省中藥綠色制造技術協同創新中心,陜西 西安 712046)
吡咯烷側鏈作為一種生物活性優異的分子片段,被廣泛應用于藥物分子的設計合成[1-3]。如已上市的用于治療骨髓纖維化的藥物Fedratinib結構中含有乙氧基吡咯烷側鏈[4]、酪氨酸激酶抑制劑西地尼布(Cediranib)結構中含有丙氧基吡咯烷側鏈[5],另外,還有具有抑制酪氨酸激酶等活性的多靶點激酶抑制劑TG100801和TG100572[6-7]、G9a組蛋白甲基轉移酶選擇性抑制劑A366和UNC0638[8-9]以及白三烯A4水解酶抑制劑SC-22716等[10]結構中均含有吡咯烷側鏈。
為了研究5-甲氧基-2-硝基-4-(3-(吡咯烷-1-基)丙氧基)苯甲酸甲酯在構筑藥物分子中的應用,作者以4-(芐氧基)-5-甲氧基-2-硝基苯甲酸甲酯(Ⅱ)為原料,經過脫芐基、羥烷基化兩步反應合成得到含有吡咯烷側鏈的目標化合物5-甲氧基-2-硝基-4-(3-(吡咯烷-1-基)丙氧基)苯甲酸甲酯(Ⅰ),通過1HNMR和ESI-MS對中間體及目標化合物的結構進行表征,并對羥烷基化條件進行優化。合成路線如圖1所示。

圖1 目標化合物的合成路線Fig.1 Synthetic route of target compound
1 實驗
1.1 試劑與儀器
4-(芐氧基)-5-甲氧基-2-硝基苯甲酸甲酯、1-(3-氯丙基)吡咯烷鹽酸鹽,泰坦科技股份有限公司;柱層析硅膠(300~400目),青島海洋化工廠;其它試劑均為市售分析純。
AV 300MHz型核磁共振波譜儀(DMSO-d6或CD3OD為溶劑,TMS為內標),德國Bruker公司;Ultima Global Spectrometer型質譜儀(ESI源),美國Waters公司;RE-52AA型旋轉蒸發儀,上海亞榮生化儀器廠;SHB-Ⅲ型循環水式多用真空泵,鄭州長城科工貿有限公司。
1.2 合成方法
1.2.1 4-羥基-5-甲氧基-2-硝基苯甲酸甲酯(Ⅲ)的合成
將4-(芐氧基)-5-甲氧基-2-硝基苯甲酸甲酯(Ⅱ)3.17 g(10.0 mmol)和三氟乙酸(25 mL)混合均勻,室溫反應4 h;TLC監測反應結束后,減壓濃縮,用甲基叔丁基醚打漿洗滌;再減壓抽濾,得灰白色固體化合物Ⅲ2.24 g,收率98.5%。1HNMR(300 MHz,DMSO-d6),δ:10.73(br,1H),7.41(s,1H),7.28(s,1H),3.91(s,3H),3.81(s,3H);ESI-MS,m/z:228.0[M+1]+。
1.2.2 目標化合物(Ⅰ)的合成
取化合物Ⅲ 1.14 g(5.0 mmol)、乙腈30 mL、1-(3-氯丙基)吡咯烷鹽酸鹽(Ⅳ)1.01 g(5.5 mmol)、 碳酸銫 3.91 g(12.0 mmol)和碘化鉀0.91 g(5.0 mmol),混合均勻,回流反應2 h;TLC監測反應結束后,減壓抽濾,濾液濃縮得到粗品;經硅膠柱層析分離純化(洗脫劑為V石油醚∶V乙酸乙酯=10∶1),得黃色黏稠狀液體化合物Ⅰ 1.41 g,收率83.3%。1HNMR(300 MHz,CD3OD),δ:7.58(s,1H),7.25(s,1H),4.18(t,J=6.3 Hz,2H),3.96(s,3H),3.89(s,3H),2.73~2.68(m,2H),2.63~2.59(m,4H),2.13~2.04(m,2H),1.89~1.80(m,4H);ESI-MS,m/z:339.1[M+1]+。
2 結果與討論
2.1 脫芐基合成化合物Ⅲ
實驗室脫芐基的方法較多,常見的有Pd/C加氫脫除法、甲酸銨/Pd/C脫除法、酸性條件脫除法等[11-14]。本實驗用三氟乙酸脫除化合物Ⅱ上的芐基合成化合物Ⅲ。具體過程:將化合物Ⅱ加入到三氟乙酸中,室溫攪拌4 h,TLC監測反應完全后減壓濃縮得到粗品,用甲基叔丁基醚打漿洗滌,再減壓抽濾,得到較高收率的化合物Ⅲ,HPLC純度達到97%。該脫芐基方法無需加熱,而且后處理操作簡單。
2.2 羥烷基化合成目標化合物
在堿性條件下,碘化鉀催化化合物Ⅲ與1-(3-氯丙基)吡咯烷鹽酸鹽(Ⅳ)的羥烷基化反應合成目標化合物。物料比(化合物Ⅳ與化合物Ⅲ物質的量比)、堿的種類和用量、反應溫度、反應時間對目標化合物的收率影響很大。
2.2.1 物料比對目標化合物收率的影響
考慮到原料成本,選擇化合物Ⅳ過量。以乙腈為反應溶劑,固定碳酸銫用量為12.0 mmol、碘化鉀用量為5.0 mmol、反應時間為2 h,考察物料比對目標化合物收率的影響,結果見表1。
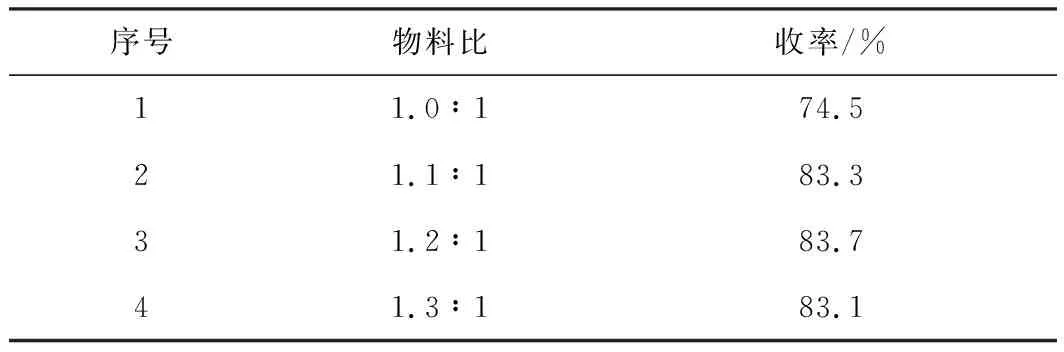
表1 物料比對目標化合物收率的影響
由表1可知,當物料比為1.0∶1時,目標化合物的收率為74.5%;增加化合物Ⅳ的用量,當物料比為1.1∶1時,目標化合物的收率達到83.3%;繼續增加化合物Ⅳ的用量,收率基本沒有變化。因此,選擇物料比n(Ⅳ)∶n(Ⅲ)為 1.1∶1。
2.2.2 堿的種類和用量對目標化合物收率的影響
化合物Ⅳ與化合物Ⅲ發生羥烷基化反應合成目標化合物的過程中,堿的作用一方面是中和化合物Ⅳ的鹽酸鹽,另一方面是使化合物Ⅲ的羥基離子化而形成鈉鹽,增強其親核性。所以,堿的種類和用量對目標化合物收率的影響很大。以乙腈為反應溶劑,固定物料比n(Ⅳ)∶n(Ⅲ)為1.3∶1、碘化鉀用量為5.0 mmol、反應時間為2 h,分別以Na2CO3、K2CO3、Cs2CO3作為反應用堿,考察堿的種類和用量對目標化合物收率的影響,結果見表2。

表2 堿的種類和用量對目標化合物收率的影響
由表2可知,分別以Na2CO3、K2CO3、Cs2CO3作為反應用堿,相同用量(10.0 mmol)下,以Cs2CO3作為反應用堿時的收率最高,達到66.0%;以Cs2CO3作為反應用堿,目標化合物的收率隨著Cs2CO3用量的增加逐漸升高,在Cs2CO3用量增至12.0 mmol時,收率達到最高,為83.3%,繼續增加Cs2CO3用量,收率沒有明顯變化。因此,選擇Cs2CO3用量為12.0 mmol。
2.2.3 反應溫度和反應時間對目標化合物收率的影響
以乙腈為反應溶劑,固定物料比n(Ⅳ)∶n(Ⅲ)為1.1∶1、碳酸銫用量為12.0 mmol、碘化鉀用量為5.0 mmol,考察反應溫度和反應時間對目標化合物收率的影響,結果見表3。

表3 反應溫度和反應時間對目標化合物收率的影響
由表3可知:(1)固定反應時間為2 h時,隨著反應溫度的升高,目標化合物的收率明顯提高,在反應溫度升至85 ℃(乙腈回流溫度)時,收率達到最高,為83.3%。(2)固定反應溫度為85 ℃時,當縮短反應時間為1 h時,收率降至68.1%;當延長反應時間至3 h時,收率基本沒有變化。因此,確定反應溫度為85 ℃、反應時間為2 h。
2.3 目標化合物的結構表征
2.3.11HNMR分析(圖2)
由圖2可以看出,δ7.579、δ7.247處的兩個單峰,積分均為1H,屬于苯環氫;δ4.181處的三重峰,積分為2H,屬于側鏈上與氧相連接的亞甲基,根據n+1規則,與之相鄰碳上為兩個氫,所以裂分為三重峰;δ3.963處的單峰,積分為3H,是甲酯上的甲基;δ3.889處的單峰,積分為3H,是苯環上的甲氧基;δ2.731~2.680處的多重峰,積分為2H,是與吡咯相連接的亞甲基;δ2.629~2.593處的多重峰,積分為4H,是吡咯上N原子相鄰的兩個亞甲基;δ2.127~2.035處的多重峰,積分為2H,是側鏈中間位置的亞甲基;δ1.894~1.800處的多重峰,積分為4H,是吡咯結構沒有與N原子相鄰的兩個亞甲基。

圖2 目標化合物的1HNMR圖譜Fig.2 1HNMR spectrum of target compound
2.3.2 ESI-MS分析(圖3)

圖3 目標化合物的ESI-MS圖譜Fig.3 ESI-MS spectrum of target compound
結合1HNMR和ESI-MS分析,進一步證明了目標化合物結構的正確性。
3 結論
以4-(芐氧基)-5-甲氧基-2-硝基苯甲酸甲酯為原料,首先在三氟乙酸條件下脫芐基得到4-羥基-5-甲氧基-2-硝基苯甲酸甲酯(Ⅲ);然后在堿性條件下與1-(3-氯丙基)吡咯烷鹽酸鹽(Ⅳ)發生羥烷基化反應得到目標化合物。脫芐基反應結束后經過簡單洗滌即可得到化合物Ⅲ。羥烷基化反應最佳條件為:乙腈為反應溶劑、物料比n(Ⅳ)∶n(Ⅲ)為 1.1∶1、碳酸銫用量12.0 mmol、碘化鉀用量5.0 mmol、反應溫度85 ℃、反應時間2 h,在該條件下,目標化合物收率達到83.3%。該研究為吡咯烷側鏈化合物的合成提供了一種可行的方案。
