超高壓液相色譜-高分辨質譜法檢測單叢烏龍茶中氨基酸
2020-11-20 03:44:50王忠合李曉婷胡文梅王軍
食品與發酵工業 2020年21期
王忠合,李曉婷,胡文梅,王軍
(韓山師范學院 食品工程與生物科技學院,廣東 潮州,521041)
單叢烏龍茶是從國家級優良品種鳳凰水仙選育出的優異單株(株系),屬于極其寶貴的品種資源,種植歷史悠久,主產于廣東省潮州市鳳凰山,是全國名茶之一,其成品茶品質優異,香味獨具一格[1-2]。成茶中的氨基酸與茶葉種類、加工程度等有密切關系,是影響茶營養保健功能及風味品質的關鍵化學成分之一,茶葉中含有多達26種氨基酸,含量約為2%~5%,其中茶氨酸占氨基酸總量的50%左右,約占茶葉干重的1%~3%。茶氨酸作為茶葉特有的化學成分,不僅構成茶湯鮮爽味,還能降低茶的苦澀味,在提神、鎮靜、降血壓、抗癌和減肥等方面都具有較好的功效作用[3-5]。游離氨基酸的含量和組成對茶湯的滋味、色澤有較明顯的影響,對茶湯的香氣和鮮爽度起著決定性作用[6]。由于茶原料、加工、制作方法的不同,茶氨酸及其他游離氨基酸在各類茶葉中的含量存在非常大的差異,輕度發酵的白茶和未發酵的綠茶中游離氨基酸的含量較高,白茶比深度發酵的黑茶中的含量高約90倍[7-8],這與不同種類的茶特有的口感和風味密切相關。因而準確測定茶葉中各游離氨基酸的含量,對于評定其營養價值和風味特性具有重要意義。
高效液相色譜法用于測定食品中氨基酸的含量已進行了大量的研究,由于大多數氨基酸無紫外吸收及熒光發射特性,需要用質譜直接檢測,也可采用衍生化后用紫外吸收或熒光檢測器檢測,常用的衍生劑有:6-氨基喹啉-N-羥基琥珀酰亞胺碳酸鹽(6-aminoquinolyl-N-hydroxysuccinimidyl carbamate,AQC)[7]、鄰苯二甲醛(o-phthalaldehyde,OPA)[9]、異硫氰酸苯酯(phenyl isothiocyanate,PITC)[10]、2,4-二硝基氟苯[11]、丹磺酰氯(dansyl chloride,dabsyl-Cl)[5]和9-芴甲氧羰酰氯(9-fluorenylmethyl chloroformate,FMOC-Cl)[12]等,其中OPA衍生化方法簡單、反應迅速、易實現自動化,但衍生物穩定性低,且不能與仲氨酸反應;FMOC-Cl和PITC可與一級和二級氨基酸反應,但其副產物較多進而干擾測定,且PITC衍生體系中的三乙胺試劑在質譜中的響應特別靈敏,會抑制其他物質的離子化,殘留性高、很難清洗干凈,在質譜的后續使用會出現三乙胺峰的干擾;dabsyl-Cl具有衍生迅速、樣品制備簡單、靈敏度高;AQC屬于專用衍生劑,具有衍生反應步驟簡單、所需時間短、副反應少、產物穩定等優點,且采用串聯質譜法替代紫外吸收或熒光檢測器分析衍生產物,不僅能夠利用質核比的差異區分共洗脫物,而且還能增加其檢測靈敏度[7]。而隨著高分辨質譜技術的發展,可以有效排除復雜食品基質中雜質引起的背景干擾,提高了檢測的準確性和精確度,并且與二級質譜圖庫比對,可以實現對化合物的驗證,從而實現快速篩查和定量測定食品中氨基酸的含量[13-14],加之高分辨質譜技術在快速篩查過程中不需要標準品,因此,高分辨質譜技術在食品分析檢測領域中的應用必將進入一個飛速發展的階段。本實驗選取5種鳳凰單叢烏龍茶為研究對象,利用超高壓液相色譜-四級桿飛行時間質譜法(ultra-high pressure liquid chromatography-quadrupole time-of-flight mass spectrometry,UHPLC-QTOF MS)測定游離氨基酸的含量,為單叢烏龍茶中游離氨基酸的準確鑒別和定量測定提供參考。
1 材料與方法
1.1 材料與試劑
單叢烏龍成茶,于2018年4月采購于廣東省潮州市鳳凰山茶園;谷氨酸(Glu)、異亮氨酸(Ile)、精氨酸(Arg)、天冬氨酸(Asp)、天冬酰胺(Asn)、谷氨酰胺(Gln)、蛋氨酸(Met)、酪氨酸(Tyr)、亮氨酸(Leu)、苯丙氨酸(Phe)、賴氨酸(Lys)、色氨酸(Trp)、丙氨酸(Ala)、甘氨酸(Gly)、蘇氨酸(Thr)、脯氨酸(Pro)、組氨酸(His)、絲氨酸(Ser)、半胱氨酸(Cys)、纈氨酸( al)、反式-4-羥基-脯氨酸(Hpro)等,Sigma-Aldrich公司;茶氨酸(The)、茚三酮,生工生物工程(上海)有限公司;質譜級乙腈和甲醇,Fisher公司;KCl、KH2PO4、K2HPO4、濃鹽酸、三乙胺、無水乙酸鈉、正己烷、冰乙酸、考馬斯亮藍G-250、體積分數95%乙醇等均為分析純;超純水(電阻率≥18.0 MΩ·cm),實驗室自制。
氨基酸混合標準溶液的配制:分別稱取22種氨基酸標準品各1.0 mg,用體積分數0.1%甲酸水溶液溶解,配成100 mg/L的儲備液。吸取一定量的氨基酸混合標準儲備液,用等體積比的乙腈-水(含0.1%甲酸)溶液稀釋配成一系列質量濃度的標準工作液,過0.22 μm濾膜,備用。
1.2 儀器與設備
Agilent 1260 Infinity超高壓液相色譜儀,由二元泵、柱溫箱、自動進樣器、脫氣機、紫外檢測器組成,安捷倫有限公司;Xe o G2-XS QTOF高分辨質譜儀,配電噴霧離子源(ESI),沃特世有限公司;JY3002電子天平,梅特勒-托利多儀器有限公司;DZF-6050型真空干燥箱,鞏義市予華儀器有限責任公司;ZX4漩渦振蕩器,意大利 ELP公司;JSP-200型高速多功能粉碎機,浙江省永康市金穗機械制造廠;WFJ 7200可見光分光光度計,尤尼柯(上海)儀器有限公司;GWA-UN1-10超純水器,北京普析通用儀器有限責任公司。
1.3 實驗方法
1.3.1 茶葉樣品處理及氨基酸的提取
分別將茶葉樣品置于干燥粉碎機中,粉碎過40目篩,-20 ℃冷藏,備用。稱取3.0 g磨碎樣品于500 mL藍蓋瓶中,加入沸蒸餾水100 mL,立即移入沸水浴中浸提20 min,浸提后立即趁熱減壓過濾,將濾液冷卻后轉入100 mL容量瓶中,用水定容至刻度,搖勻,待用。
1.3.2 比色法測定氨基酸的總量
采用GB/T 8314—2013《茶 游離氨基酸總量的測定》[15]測定提取液中氨基酸的總量,準確吸取1.0 mL樣品提取液或標準工作液,注入25 mL比色管中,加入0.5 mL pH 8.0磷酸鹽緩沖液和0.5 mL 20 mg/mL茚三酮溶液,在沸水浴中加熱15 min。待冷卻后加水定容至25 mL。放置10 min后,在570 nm處,以試劑空白溶液作參比,測定吸光度。分別根據標準曲線方程Y=0.0612X-0.0234(R2=0.9967)求得氨基酸的質量濃度,代入公式(1)計算出樣品中總氨基酸的含量:
式中:ω,氨基酸的含量,mg/kg;ρ,由標曲求得的質量濃度,mg/L;,樣液定容體積,mL;m,取樣質量,g。
1.3.3 超高壓液相色譜-高分辨質譜法測定各氨基酸含量
取氨基酸標準液或樣品上清液分別過0.22 μm濾膜后采用UHPLC-QTOF MS進行檢測,根據氨基酸標準曲線,計算出茶葉中各游離氨基酸的含量。色譜條件[16-17]:HILIC-Z色譜柱(2.1 mm×100 mm,2.7 μm);流速 0.4 mL/min;柱溫 30.0 ℃;進樣量 2 μL;洗針進樣,流動相A:20 mmol/L甲酸銨水溶液(含體積分數0.1%甲酸)、流動相B:乙腈(含體積分數0.1%甲酸),梯度洗脫(優化后的程序如表1)。質譜條件:正離子靈敏度模式檢測,掃描范圍m/z50~600,毛細管電壓2.63 k ,錐孔電壓40 ,提取錐孔電壓6 ,離子源溫度130 ℃;脫溶劑氣溫度400 ℃;錐孔氣流速60 L/h,脫溶劑氣流速800 L/h,掃描時間0.2 s。全掃描質譜數據(MS)采集方法:第1通道碰撞能量6 e 、第2通道碰撞能量15、25和35 e 。全信息質譜數據(MSE)采集方法:低能通道碰撞能量6 e 、高能通道碰撞能量從10增至35 e 。2種采集模式下的數據類型均為棒狀圖、采用質量鎖定技術校正以獲取準確有效的質量數測量值,以200 pg/μL的亮氨酸腦啡肽溶液為鎖定質量溶液,流速為10 μL/min,電壓為2.0 k ,每隔30 s采集1次,在正離子模式下產生m/z556.277 1的離子。

表1 梯度洗脫程序表Table 1 Gradient elution program employed for the separation of amino acids
1.3.4 精密度實驗
精密量取2.0 mL氨基酸標準工作溶液置于10 mL容量瓶中,加體積比為1∶1的乙腈-水溶液(含體積分數0.1%單酸)稀釋至刻度,搖勻,制成標準混合溶液,連續進樣6次,計算氨基酸的峰面積和相對標準偏差。
1.3.5 加標回收實驗
稱取單叢烏龍茶葉樣品9份,分別提取后置10 mL容量瓶中,精密加入2.0 mL氨基酸的混合標準工作溶液低、中、高3個質量濃度水平(1、10、50倍定量限),加適量體積比為1∶1的乙腈-水溶液(含體積分數0.1%單酸),超聲處理15 min,取出,冷卻至室溫,再稀釋至刻度,搖勻,分別作為低、中、高3個質量濃度回收率組,每個質量濃度水平配制3 份,配制的溶液過0.22 μm微孔濾膜,注入超高壓液相色譜串聯高分辨質譜儀測定。
1.4 數據處理
采用Waters公司的MassLynx 4.1軟件,通過提取離子峰獲得化合物的色譜圖并選用ApexTrack的積分方法,適當調整積分參數的方式完成色譜積分,根據質譜圖中碎片離子峰的識別與峰匹配情況確認化合物。實驗結果以平均值±標準偏差表示,采用SPSS 17.0進行單因素方差分析,并采用鄧肯檢驗進行差異顯著性分析,以P<0.05為檢驗標準。
2 結果與討論
2.1 質譜參數的優化
毛細管電壓、錐孔電壓和碰撞能量是影響質譜測定結果的重要因素,錐孔電壓直接影響各組分的靈敏度[11],過高的錐孔電壓會導致分子離子在離子源內發生碰撞解離,影響母離子的豐度,進而影響檢測限和靈敏度。本文采用的MSE和MS兩種質譜采集模式均可采集母離子和碎片離子,但不同采集模式對準分子離子峰和碎片離子峰的豐度及數量的影響較大,將氨基酸標樣混合溶液注入質譜儀,優化質譜參數,以獲得最優的靈敏度和較多的碎片離子,且豐度值較高,如茶氨酸在不同質譜采集模式下的結果如圖1所示。
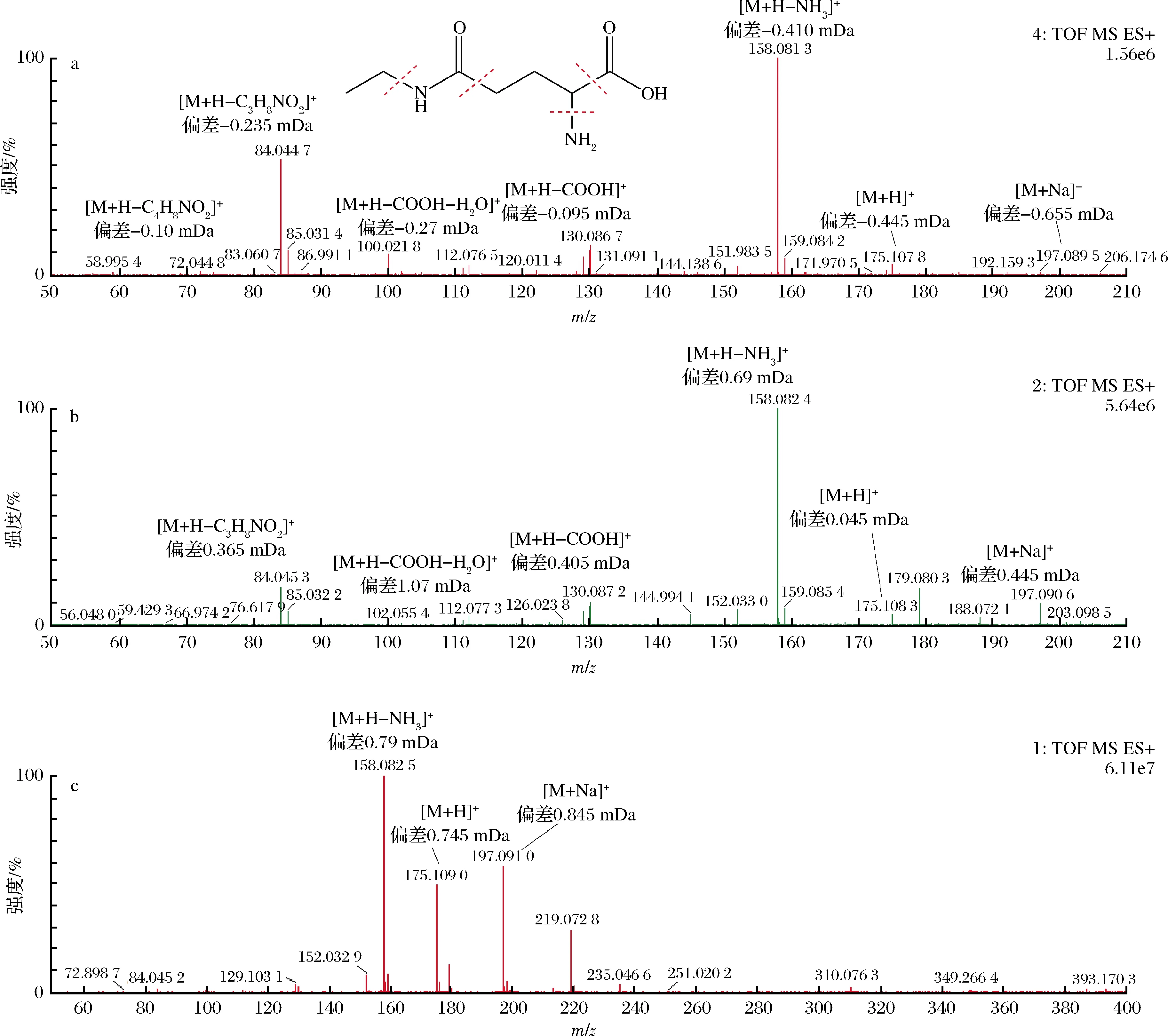
a-MS采集模式;b-MSE采集模式;c-茶氨酸碎片離子和母離子圖1 不同采集模式MS和MSE下的茶氨酸碎片離子和母離子質譜圖Fig.1 Mass spectrograms of theanine fragment and parent ions in MS and MSE acquisition modes
MSE采集模式是利用低能通道和高能通道2種掃描快速切換交替構成,分別記錄母離子及碎片離子信息,可以進行鑒別和定量分析,但此模式的高能通道中碰撞能量只能設定范圍值。MS采集模式也是采用低能通道掃描獲得母離子信息,而高能通道的碰撞能量則有多種設置方式,如:固定值、一組值或范圍值等。由圖1可知,茶氨酸的一級質譜圖中主要有加合H+、加合Na+的準分子離子峰和脫去氨的離子峰,且脫氨的離子峰的豐度最高,用于定量分析可提高檢測限和靈敏度[17],其他氨基酸的定量離子峰如表2。而在MS(碰撞能15 )和MSE(碰撞能10~35 )2種不同采集模式下獲得的茶氨酸高能通道質譜圖中各碎片離子的數量和豐度差異較大,MS采集模式中共獲得7個準分子離子峰和碎片離子峰,準分子離子峰的豐度較低,大多數的準分子離子在碰撞中變成碎片離子,故在m/z84.044 7處的豐度較高;而MSE采集模式中可獲得9個準分子離子峰和碎片離子峰,準分子離子峰的豐度較清晰,碎片離子峰的豐度較低,但在m/z158.082 4處的離子峰豐度最高,該采集模式獲得的碎片離子信息多更適合于鑒別茶葉中的各種氨基酸,因而后續研究中質譜采用MSE模式采集。
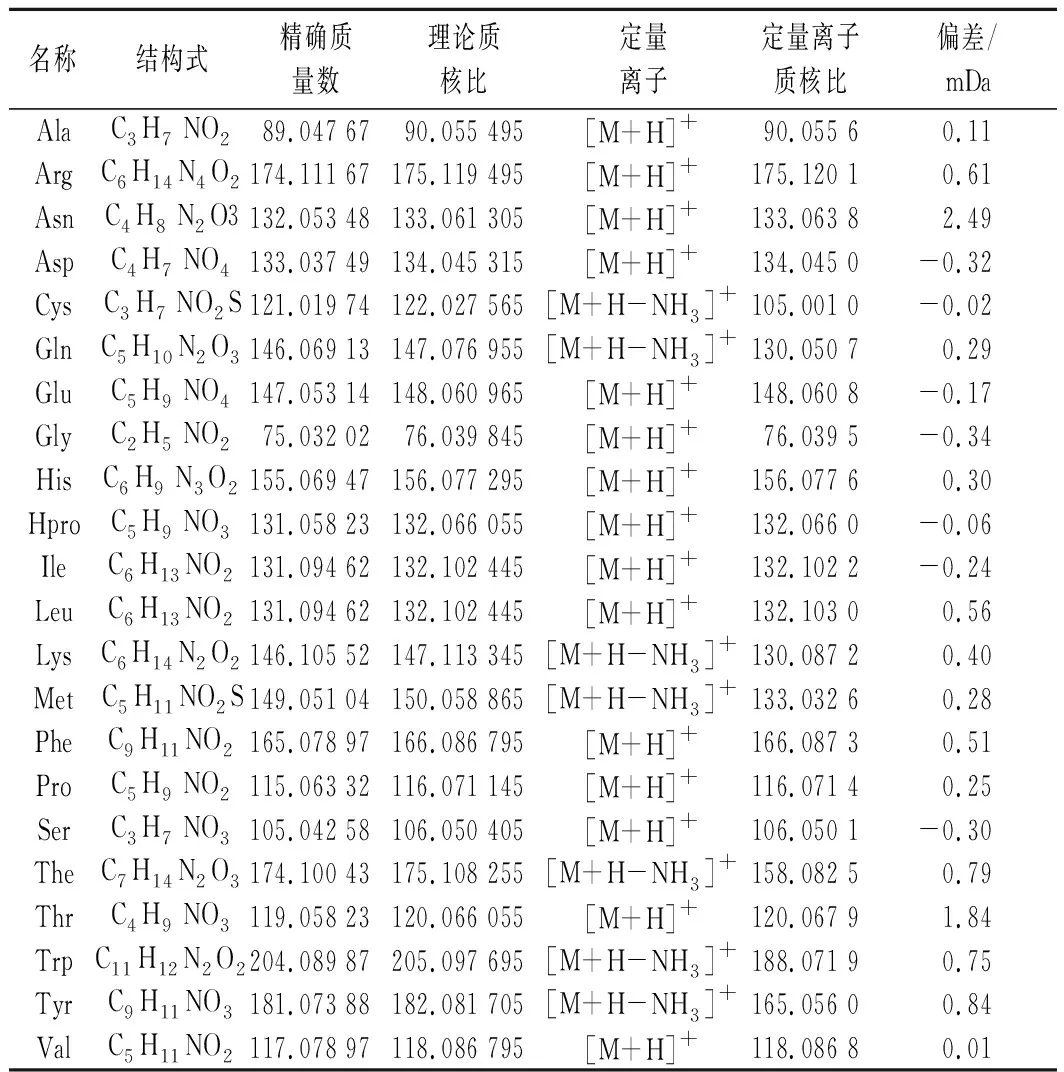
表2 氨基酸標準品的精確質量及相關數據Table 2 Monoisotopic mass and related data of standard amino acids
2.2 色譜參數的優化
實驗初期,分別采用了SB-C18柱(250 mm×4. 6 mm,5.0 μm)、EC-C18柱(100 mm×2. 1 mm,2.7 μm)和HILIC-Z柱(100 mm×2.1 mm,2.7 μm),在二元流動相為甲酸銨-甲酸水溶液-乙腈的梯度洗脫條件下,對22種氨基酸的混合標準溶液進行了分離條件的優化。C18柱的分離效果明顯不如HILIC-Z柱,部分氨基酸的出峰時間重合無法分離,如賴氨酸、精氨酸和組氨酸的峰重合,這主要是由于3種氨基酸的水溶性較好,在C18柱上的保留差,分離效果較差。亮氨酸和異亮氨酸在C18柱上也比較難實現分離,由于這2種氨基酸具有相同的質核比,因而無法進行鑒別和定量分析[13]。鑒于此,優化了HILIC-Z柱分離氨基酸的色譜條件,采用同樣的二元流動相進行梯度洗脫優化,如圖2所示,氨基酸標準化合物的分離效果理想,亮氨酸和異亮氨酸可分開,且分離時間只需15 min,降低了分析所需時間和溶劑的消耗等,能夠達到快速、高效分離的要求,且該分離條件不需添加三乙胺和難揮發性的無機鹽,只用到揮發性的甲酸和甲酸銨改性劑,非常適合于質譜檢測,優化后的梯度洗脫條件如表1所示。

2.3 方法學考察
移取適量的氨基酸混合標準工作溶液,加入到單叢烏龍茶樣品粉末中,按照1.3.1小節的方法處理,配制系列質量濃度的氨基酸混合標準溶液測定,各氨基酸的檢測結果如表3所示。依據各氨基酸的定量離子峰(如表2)提取的色譜圖中信噪比(S/N)大于3和10得到檢測限與定量限,22種目標物的線性相關系數為0.99~0.9996,樣品中各測定出的氨基酸化合物均在線性范圍內,檢測限為1.3~95.7 μg/L。

表3 氨基酸標準品的保留時間、標準曲線方程、檢測限及定量限Table 3 Retention time, standard cur e equation, limits of detection (LOD) and limits of quantitation (LOQ) of standard amino acids
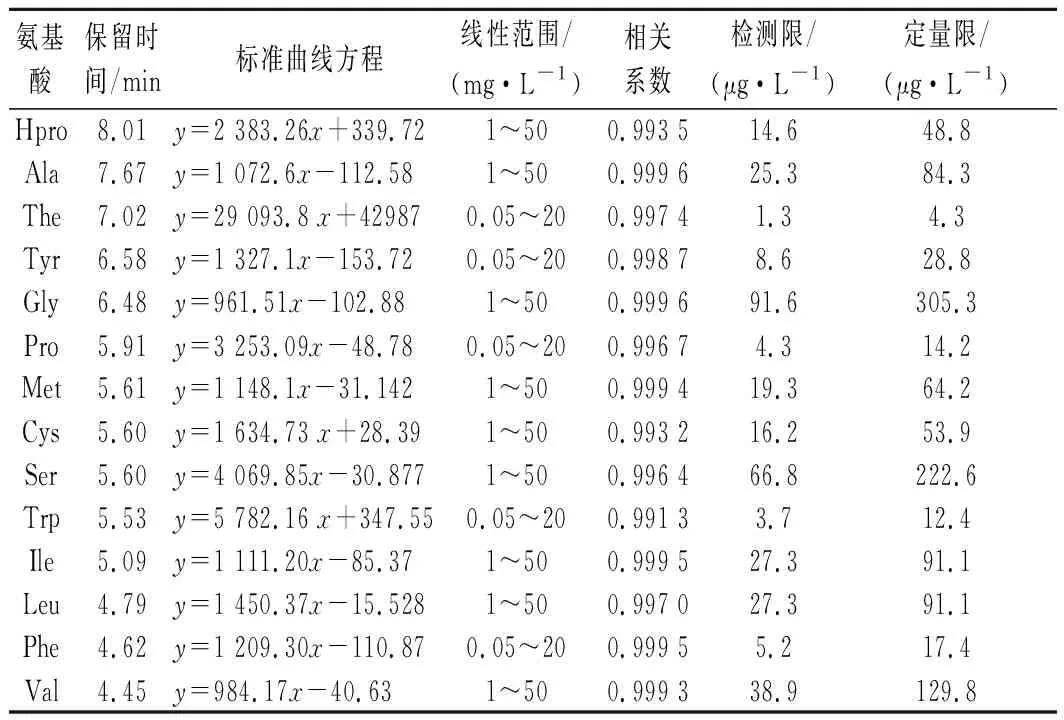
續表3
移取適量的氨基酸混合標準工作溶液,加入到單叢烏龍茶樣品粉末中,配制低、中、高3個質量濃度水平(1、10、50倍定量限)的添加試驗,各平行5份,按照1.3.1小節的方法處理,回收率和精密度實驗測定結果如表4所示。由表4可知,各氨基酸目標物的加標回收率為83%~112%,相對標準偏差小于5%,可用于單叢茶樣品中氨基酸含量的測定。
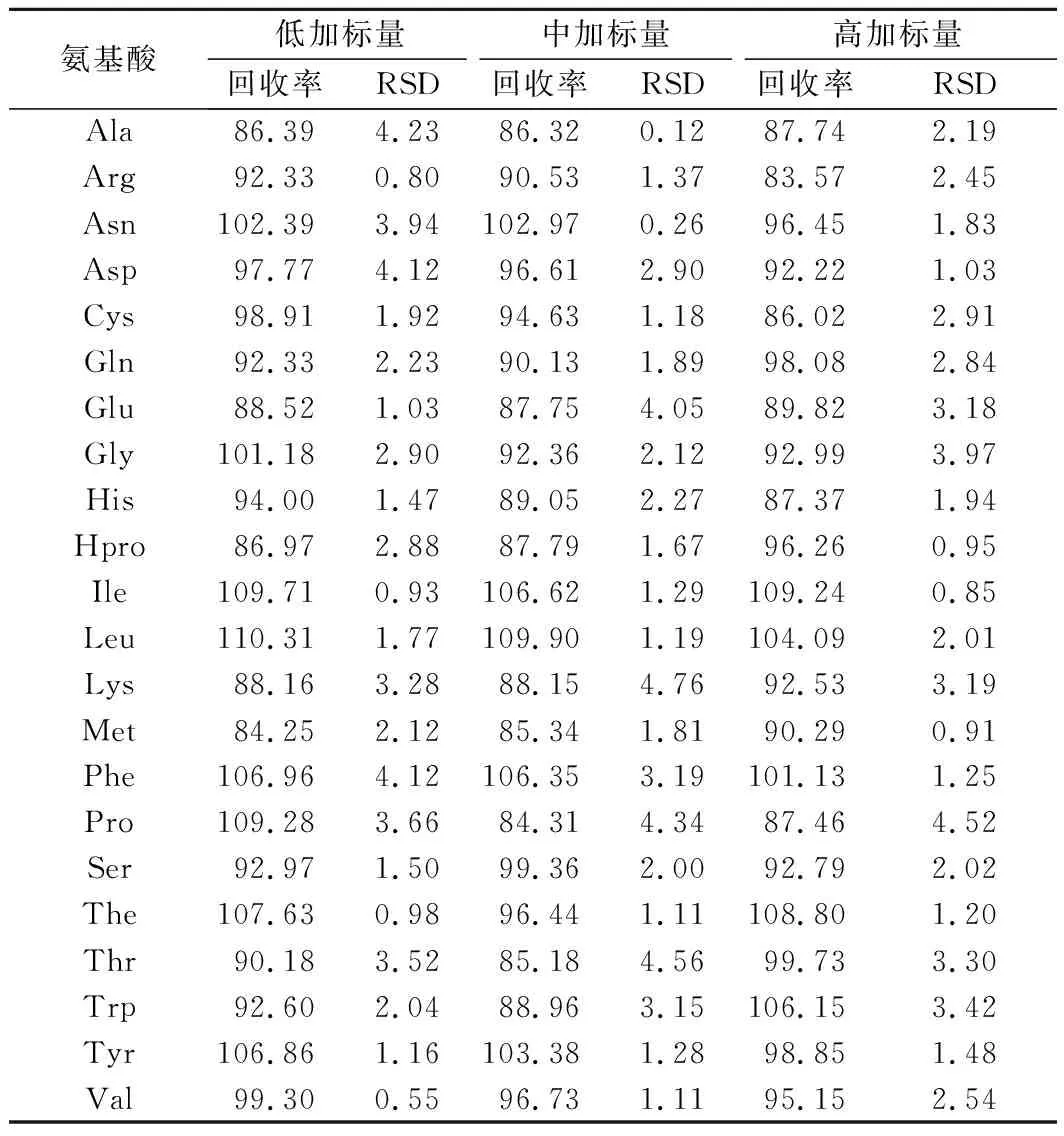
表4 單叢烏龍茶樣品中氨基酸測定的添加回收率及其相對標準偏差(RSD) 單位:%
2.4 茶葉樣品中氨基酸鑒別與含量測定
圖3為單叢茶樣品測定的總離子流圖和不同質量窗口提取的茶氨酸和精氨酸的準分子離子色譜圖。提高質量數測定的準確度,有利于提高篩查鑒別的可靠性,減少假陽性結果出現的可能性。串聯質譜可區分質核比的精度只有0.01 Da,借助碎片離子可定量測定和區分化合物,但對于共流出的質核比相近的碎片則無能為力。而高分辨質譜則可精確到0.1 mDa,再根據碎片離子質譜信息和同位素豐度比值等信息可對化合物進行準確篩查和確證。如圖3-b所示,在提取質量窗口為0.05 Da下提取茶氨酸(m/z175.108)和精氨酸(m/z175.120)的色譜圖中均在6.83 min會出現同1個色譜峰,而當提取質量窗口為0.005 Da下提取精氨酸(m/z175.120)的色譜圖中不會出現干擾峰,可準確提取到精氨酸的色譜峰。茶氨酸和精氨酸的平均相對分子質量為174.197 7和174.201,2種化合物失去1個氨基(m/z17)和1個羧基(m/z45)得到的碎片離子峰也相同[18],因而串聯質譜的質量精度無法區分這兩種氨基酸,已報道的茶氨酸測定方法中未考慮精氨酸的影響,m/z158的峰推測錯誤[19]。由表2可知,茶氨酸和精氨酸的單同位素精確質量數為174.111 67和174.100 43,兩者相差約11 mDa,只有用高分辨質譜才能準確區分。由此可見,采用高質量的色譜圖可減少假陽性的篩查結果,獲得清晰信噪比高的色譜圖,有利于提高檢出限。

a-總離子流圖;b-色譜圖圖3 單叢烏龍茶樣品測定的總離子流圖及不同質量窗口下的茶氨酸及精氨酸提取色譜圖Fig.3 Total ion chromatogram of Oolong tea extract and extracted ion chromatogram of arginine and theanine in different mass chromatogram window
高分辨質譜法可以提供化合物的保留時間,質譜碎片離子、精確質量數、同位素豐度比值等信息,這些信息可為化合物的準確定性和篩查鑒別提供充分的依據。在實驗中根據氨基酸標準化合物的碎片離子信息確定其在茶葉樣品中的存在情況,并根據提取色譜圖的峰面積進行定量,結果如表5所示。

表5 單叢烏龍成茶中氨基酸種類與含量 單位:mg/100 g
由表5可知,采用UHPLC-QTOF法測定的5種單叢烏龍成茶中游離氨基酸的種類相同,主要有茶氨酸、谷氨酸、精氨酸、絲氨酸、天冬氨酸等,但各氨基酸在不同單叢烏龍成茶中的含量差異較大(P<0.05),不同茶葉中各游離氨基酸的加和值相差較大,這可能與茶樹品種、生長環境、加工條件等因素密切相關,如玉蘭香茶葉中茶氨酸和天冬氨酸的含量較高,而谷氨酸的含量較低。采用茚三酮比色法測定的5種單叢烏龍成茶中氨基酸總量差異顯著(P<0.05),且與各游離氨基酸的加和值差異較大,這可能是由于茶葉提取液中含有其他氨基化合物,因而茚三酮比色法測定的氨基酸總量值偏大,這進一步表明不同成茶中游離氨基酸種類和含量有較大差異,與其種類和加工過程密切相關。晾青過程中在酶的作用下蛋白質水解的游離氨基酸含量增加,這與加工過程中內源性蛋白酶被激活,蛋白質在各種蛋白酶的催化作用下水解成多種游離氨基酸,使游離氨基酸的總量增加[7],從分子和蛋白水平看,茶葉晾青過程中茶氨酸的生物代謝與CsFd-GOGAT和CsNADH-GOGAT基因的表達密切相關[20]。發酵過程中游離氨基酸可發生氧化脫氨或脫羧反應形成芳香物質,也可通過羰氨反應產生香氣成分,從而使得游離氨基酸的含量降低,其中茶氨酸作為一種茶樹特征性非蛋白主體氨基酸可與兒茶素結合形成黃酮堿類物質,這與烏龍茶獨特香味形成有密切聯系,因而在不同的成茶中其含量差異較大。
3 結論
HILIC柱分離單叢烏龍成茶中氨基酸的效果理想,亮氨酸和異亮氨酸在15 min內即可分開,該分離條件不需添加三乙胺和難揮發的鹽,非常適合用于質譜檢測。質譜采集模式對準分子離子峰和碎片離子峰的豐度及數量的影響較大,在電噴霧正離子化(ESI+)檢測,22種氨基酸目標物的線性相關系數為0.99~0.9996,檢出限為1.3~66.8 μg/L,加標回收率為83%~112%,相對標準偏差均小于5%,可用于單叢烏龍茶樣品中氨基酸含量的測定。
茚三酮比色法測定5種單叢茶樣品中總游離氨基酸的總量為485.12~575.95 mg/100 g,差異顯著(P<0.05)。超高壓液相色譜-高分辨質譜法測定結果表明,5種單叢烏龍成茶樣品中游離氨基酸的種類相同,主要有茶氨酸、谷氨酸、精氨酸、絲氨酸、天冬氨酸等,但各氨基酸在不同單叢烏龍成茶中的含量差異較大(P<0.05),這與不同品種的單叢烏龍茶獨特的香味形成及口感有密切聯系。
