HPLC-MS/MS法測定腌制食品中色素含量的預處理方法比較
2020-11-24 06:26:04鐘萍陳鮮麗羅威李亞楠
食品研究與開發 2020年21期
關鍵詞:效應
鐘萍,陳鮮麗,羅威,*,李亞楠
(1.湛江幼兒師范專科學校,廣東湛江524084;2.嶺南師范學院基礎教育學院,廣東湛江524037;3.韶關學院醫學院,廣東韶關512026)
蘇丹紅I~IV、蘇丹紅G、蘇丹橙G和蘇丹紅7B均為合成色素,既可用于石油、機油等化工產品的調色,也可用于皮革、紡織品或家具的涂染[1-2]。由于上述化合物的結構中均有偶氮結構,人體若長期接觸,會對肝、腎臟器產生強烈毒性作用,并有致癌風險[3-4],因此國內外食品安全監管部門明確要求,不允許將其作為食品添加劑使用,因此建立高效、靈敏、準確的檢測方法,對相關食品的安全監管意義重大。
目前蘇丹紅I~IV、蘇丹紅G、蘇丹橙G和蘇丹紅7B的檢測方法主要有紫外分光光度法[5]、薄層色譜法[6]、高效液相色譜法(high performance liquid chromatography,HPLC)[7-8]、高效液相色譜-質譜法(high performance liquid chromatography-tandem mass spectrometry,HPLC-MS)[9-10]、氣相色譜法[11]及酶聯免疫檢測法[12]等。其中以HPLC法和HPLC-MS/MS法應用最為常見,但高效液相色譜法的靈敏度較低,且易受共存組分的干擾,而其與質譜聯用后,可克服上述缺點,具有靈敏度高、選擇性強且定性與定量準確等特點,利于排除液相色譜的假陽性結果,因而利用HPLCMS/MS法檢測腌制肉類食品中合成色素的研究被廣泛報道[13-15],但不同預處理樣品方法對測定結果的影響,卻鮮有提及。因此,本研究分別采用液液萃取-凝膠滲透色譜(liquid liquid extraction-gel permeation chromatography,LLE-GPC)法、固相萃取法(solid-phase extraction,SPE)和QuEChERS法預處理腌制肉類食品后,利用HPLC-MS/MS法測定合成色素的含量,從而為相關食品中合成色素的定量檢測提供參考。
1 材料與方法
1.1 儀器與試劑
蘇丹紅I、蘇丹紅II、蘇丹紅III、蘇丹紅IV、蘇丹紅G、蘇丹橙G和蘇丹紅7B標準物質(純度≥90.0%):上海安譜科學儀器有限公司;臘腸:市售;甲醇、乙腈、甲酸、醋酸銨、乙酸乙酯、環己烷、正己烷、二氯甲烷均為色譜純:德國默克公司;乙酸、無水硫酸鈉均為分析純:國藥集團化學試劑有限公司;硅膠(C18)填料、N-丙基乙二胺(primary secondary amine,PSA)填料、Bio-Beads S-X3填料:天津博納艾杰爾科技有限公司;試驗用水為高純水(電阻率大于18.2 MΩ·cm)。
PL-GPC220凝膠滲透色譜儀、Agilent 1200高效液相色譜:美國安捷倫科技公司;API 4000四極桿質譜儀:美國PE公司;ME204電子天平:梅特勒-托利多公司;LG-22M高速冷凍離心機:四川蜀科儀器有限公司;FV64全自動氮吹濃縮儀:廣州得泰儀器科技有限公司;FRQ-1020超聲波清洗器:溫州百亞超聲設備有限公司;XH-D渦旋混合器上海冠森生物科技公司;HN-98IB組織勻漿機:上海達洛科學儀器有限公司。
1.2 方法
1.2.1 標準溶液配制
1.2.1.1 混合標準儲備液配制
分別準確稱取蘇丹紅I、蘇丹紅II、蘇丹紅III、蘇丹紅IV、蘇丹紅G、蘇丹橙G和蘇丹紅7B標準物質10.0 mg(精確至0.000 1 g),共置于100 mL容量瓶內,用乙腈稀釋至刻度線定容、搖勻,即得0.1 mg/mL混合標準儲備液。
1.2.1.2 混合標準溶液配制
分別精密移取上述混合標準儲備液0.10、0.50、1.0、5.0、10.0 mL,置于 100 mL 容量瓶內,用乙腈稀釋至刻度線定容、搖勻,配制成濃度為0.10、0.50、1.0、5.0、10.0 μg/mL的混合標準溶液,現用現配。
1.2.2 儀器工作條件
1.2.2.1 凝膠滲透色譜條件
凝膠色譜柱(400 mm×25 mm),填料為Bio-Beads S-X3(0.037 mm~0.075 mm);淋洗液:乙酸乙酯-環己烷(1∶1,體積比);流量:5.0 mL/min;進樣體積:5 mL。
1.2.2.2 液相色譜條件
Agilent Zorbax SB C18色譜柱(100 mm×4.6 mm,5 μm);柱溫為 30 ℃;流量為 1.0 mL/min;進樣體積10 μL;流動相:流動相A為0.1%(體積分數)甲酸、流動相 B 為乙腈;梯度洗脫程序:0~4 min 60%B;6 min~8 min時,B線性升高至100%;10 min~12 min時B線性降低至60%后,保持8 min。
1.2.2.3 質譜條件
電噴霧離子源(electrospray ionization,ESI);掃描方式為正離子掃描模式;檢測方式為多反應監測模式(multi-reaction monitoring,MRM);離子源溫度600℃;氣簾氣流量30 L/min;霧化氣流量40 L/min;輔助干燥氣流量50 L/min;噴霧電壓為5 200 V。
MRM監測模式下7種合成色素的特征離子及其它質譜參數見表1。

表1 7種合成色素的質譜參數Table 1 Optimal mass parameters for the seven kinds of synthetic colorants
1.2.3 LLE-GPC法
1.2.3.1 樣品制備
超市購置的臘腸食品,搗碎后混勻,置于組織勻漿機內,完全粉碎制得供試樣品。
1.2.3.2 待測物提取
準確稱取粉碎完全后的樣品2.0g(精確至0.0001g),置于50 mL聚四氟乙烯離心管內,精密移取20 mL乙酸乙酯-環己烷(1∶1,體積比),搖勻后渦旋振蕩15 min后,加入2 g無水硫酸鈉,繼續振蕩10 min后靜置至上層溶液澄清,離心7 min后(轉速:4 500 r/min),吸取上清液,重復上述步驟,合并兩次提取液,過0.45 μm濾膜,即得提取溶液。
1.2.3.3 GPC純化
精密移取提取溶液10 mL,采用1.2.2.1凝膠滲透色譜條件凈化1.2.3.2中提取液,收集22 min~28 min洗脫液后,于40℃氮氣濃縮蒸干,殘余物采用乙腈定容至1.0 mL,經0.22 μm濾膜過濾后,上樣至高效液相色譜串聯質譜儀中檢測。
1.2.4 SPE純化
精密移取提取溶液10 mL至活化后的Thermo SCX固相萃取柱,經50 mL正己烷淋洗去除油脂后,采用20 mL二氯甲烷洗脫,收集洗脫液,于40℃氮氣濃縮蒸干,采用乙腈定容至1 mL后,過0.22 μm濾膜,待測。
1.2.5 QuEChERS法
準確稱取粉碎完全后的樣品2.0g(精確至0.0001g),置于50 mL聚四氟乙烯離心管內,加入20 mL乙腈-水(1∶1,體積比),渦旋振蕩15 min后,加入2 g無水硫酸鈉,繼續振蕩 10 min,離心 7 min后(轉速:4 500 r/min),吸取上清液,重復上述步驟,合并兩次提取液后,精密移取10 mL,加入至裝有100 mg硅膠(C18)吸附劑的聚四氟乙烯離心管內,渦旋振蕩5 min后,離心5 min(轉速:4 500 r/min),吸取上清液,過 0.22 μm 濾膜,待測。
1.2.6 基質干擾評價
為評估不同預處理方法的試驗條件對基質干擾的去除效果,在乙腈和空白臘腸中分別加入等體積1.0μg/mL的混合標準溶液,照公式:基質效應(ME/%)=|(A/B-1)|×100,(式中:A為空白臘腸中色素的質譜響應強度;B為乙腈中色素的質譜響應強度)計算采用不同預處理方法試驗條件的基質效應。參考相關文獻評價標準[16],當ME/%<10%不存在基質干擾,10%<ME/%<20%時,為低程度干擾,20%<ME/%<50%時,為中程度干擾,ME/%>50%時,則基質嚴重干擾目標化合物的測定。
2 結果與分析
2.1 不同化合物的色譜圖
根據7種合成色素的特征離子,在MRM監測模式下不同化合物的總離子流圖,見圖1,LLE-GPC法中凝膠滲透色譜分離,見圖2。

圖1 7種合成色素的總離子流色譜圖Fig.1 The total ion chromatogram of seven kinds of synthetic colorants
2.2 線性范圍與檢出限

圖2 GPC純化加標樣品色譜圖Fig.2 The Gel permeation chromatogram
采用標準加入法,利用HPLC-MS/MS法測定不同質量濃度的混合標準溶液,以質譜響應強度(y)作縱坐標,7種合成色素的質量濃度為橫坐標(x),繪制標準曲線,同時以3倍信噪比(S/N)為檢測限,以10倍信噪比(S/N)為定量限,得到不同分析物在一定線性范圍下的回歸方程、相關系數及檢測限與定量限,結果見表2。
2.3 LLE-GPC法條件優化
2.3.1 提取溶劑選擇
提取溶劑對目標化合物的提取效率直接影響測定結果的準確度,為保證樣品中目標化合物完全富集,選擇不同提取溶劑,照1.2.3.2步驟,分別萃取1.2.6中樣品溶液,考察正己烷、環己烷、乙酸乙酯、乙酸乙酯-環己烷(1∶1,體積比)和乙酸乙酯-正己烷(1∶1,體積比)對基質效應的影響,結果發現,不同提取溶劑的ME/%均低于10%,但考慮后續凝膠色譜凈化的洗脫液為乙酸乙酯-環己烷(1∶1,體積比),因此確定乙酸乙酯-環己烷(1∶1,體積比)為提取溶劑。

表2 線性范圍、線性回歸方程、方法檢測限與定量限Table 2 Linear ranges,linear regression equation,limit of detection and limit of quantitation
2.3.2 提取次數選擇
提取次數間接反映提取效率,照1.2.3.2步驟,分別考察提取次數對基質效應的影響,結果發現,樣品溶液經兩次提取后,對不同色素的基質效應可降低至5%以下,因此確定提取次數為兩次。
2.4 SPE法條件優化
2.4.1 固相萃取柱選擇
固相萃取需利用固相萃取柱選擇性吸附目標化合物,經液相淋洗除雜后,再選擇性洗脫待測組分,從而實現待測物的分離、凈化與富集,因此選擇不同類型固相萃取柱,照1.2.3.2步驟,分別處理1.2.6中樣品溶液,考察 Dikma ProElut Silica、Agilent Bond Elut PSA、Thermo SCX固相萃取柱對基質效應的影響,結果見圖3。
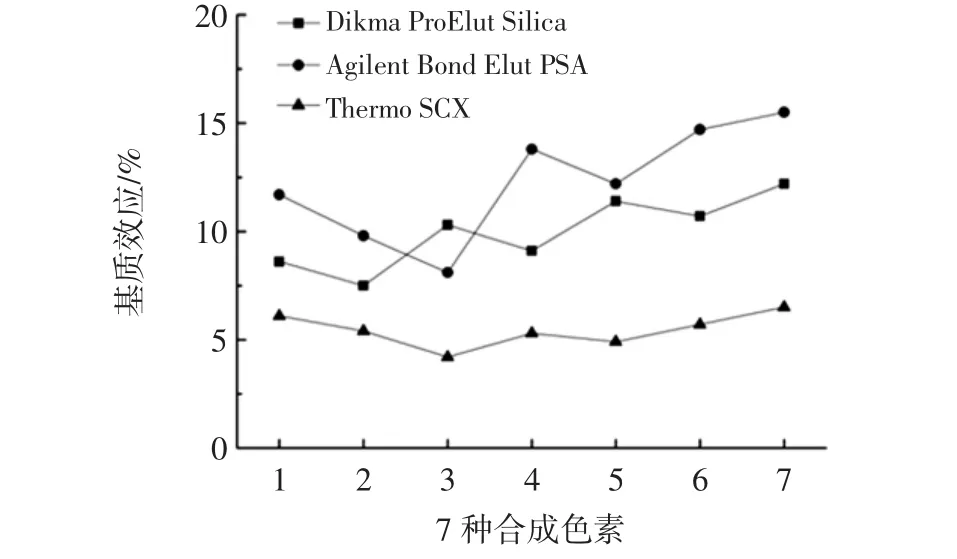
圖3 固相萃取柱對基質效應的影響Fig.3 The effect of solid phase extraction column on matrix effect
從圖3可知,相較其它兩種固相萃取柱,采用Thermo SCX固相萃取柱的基質效應可降低至10%以下,這歸因于待測7種合成色素的化學結構中均存在苯環偶氮結構,易形成季銨鹽陽離子,構成π-π共軛穩定體系,而Thermo SCX固相萃取柱的固定相為苯磺酸基,具有強陽離子交換作用[17],因此對上述合成色素均有較好保留,從而可有效去除基質干擾,因此選擇采用Thermo SCX固相萃取柱。
2.4.2 洗脫劑選擇
提取溶液上樣至固相萃取柱后,經正己烷洗滌去除油脂、蛋白質和非極性的雜質后,采用洗脫劑選擇性洗脫待測組分,因此考察等體積的不同洗脫劑(甲醇、丙酮、乙腈、二氯甲烷)對基質效應的影響,結果發現,與其它洗脫劑相比,二氯甲烷對目標化合物檢測的基質效應,可降至最低,因此采用二氯甲烷作為洗脫劑。
2.5 QuEChERS法條件優化
2.5.1 提取劑選擇
由于7種合成色素的水溶性較好,化學性質相近,因此分別選擇等體積的不同溶劑(乙腈、乙腈-水(1∶1,體積比)、水、甲醇、甲醇-水(1∶1,體積比))作為提取劑,照1.2.5步驟,預處理1.2.6中樣品溶液,考察對基質效應的影響,結果發現,當采用乙腈-水(1∶1,體積比)提取時,對7種合成色素檢測的平均基質效應最低(4.7%),這可能歸因于乙腈-水(1∶1,體積比)提取目標物和其它極性雜質后,加入無水硫酸鈉發生鹽析效應,促使乙腈與水分層,從而除去溶于水相的蛋白質、糖分等雜質[18],因此選用乙腈-水(1∶1,體積比)作為QuEChERS法預處理的提取劑。
2.5.2 吸附劑選擇
QuEChERS法常用 C18、N-丙基乙二胺(PSA)作為吸附劑,以去除提取溶劑中的糖類和蛋白質,因此分別照1.2.5步驟,預處理1.2.6中樣品溶液,考察不同種類與用量的吸附劑對基質效應的影響,結果見圖4。

圖4 吸附劑對基質效應的影響Fig.4 The effect of adsorbent on matrix effect
從圖4可知,當吸附劑為PSA時,樣品基質對合成色素檢測時干擾較小,7種色素的平均基質效應為5%左右,而C18相對較高,可能歸因于對部分色素具有一定吸附作用,另外隨著PSA用量的增多,平均基質效應降低并不明顯,因此選擇100 mg PSA作為QuEChERS法預處理的吸附劑。
2.6 不同預處理方法結果的準確度與精密度
為考察3種預處理方法結果的準確度與精密度,采用回收率表示結果準確度,相對標準偏差(relative standard deviation,RSD)表示結果的精密度,分別照1.2.3、1.2.4和1.2.5所述步驟,預處理3個水平(0.50、1.0、5.0 μg/mL)的加標樣品,進行回收試驗,每個水平平行測定5次,結果見表3。

表3 不同預處理方法的測定結果回收率與相對標準偏差(n=5)Table 3 The average recoveries and RSDs of results in different pretreatment methods(n=5)
從表3可知,SPE法的不同濃度水平的平均回收率在70%~110%之間,而QuEChERS法與LLE-GPC法的不同濃度水平的平均回收率均在90%~110%之間,優于SPE法,這可能歸因于提取溶液在固相萃取時,未被完全洗脫,3種方法的相對標準偏差均低于5%。與LLE-GPC法、SPE法相比,QuEChERS法的提取、除雜、純化過程簡便、快速,且無需昂貴的儀器與耗材,因而綜合預處理效率較高,適于相關產品的快速檢測。
3 結論
采用液液萃取-凝膠滲透色譜法、固相萃取法和QuEChERS法預處理腌制肉類食品后,利用高效液相色譜-質譜法同時測定其合成色素的含量,固相萃取法的不同濃度水平的平均回收率在80%~110%之間,而QuEChERS法與液液萃取-凝膠滲透色譜法的不同濃度水平的平均回收率均在90%~110%之間。與液液萃取-凝膠滲透色譜法、固相萃取法相較,QuEChERS法預處理樣品的操作更為簡便、快速,且處理成本適中,綜合預處理效率較高,因此適用于腌制肉類食品中合成色素的測定。
猜你喜歡
核科學與工程(2021年4期)2022-01-12 06:30:26
今日農業(2020年19期)2020-12-14 14:16:52
小學生必讀(中年級版)(2020年9期)2020-12-04 02:07:22
科學大眾(2020年17期)2020-10-27 02:49:10
紅土地(2018年11期)2018-12-19 05:10:56
意林·全彩Color(2018年9期)2018-11-13 22:49:38
中學物理·高中(2016年12期)2017-04-22 11:53:03
中國衛生(2016年4期)2016-11-12 13:24:14
中國衛生(2014年4期)2014-12-06 05:57:14
小櫻桃·童年閱讀(2014年11期)2014-12-01 22:21:30
