AuCl3基乙炔氫氯化催化劑的穩定性與催化性能研究
2020-11-27 04:45:16陳海天朱峰云孫云峰申玉海黃太仲
山東化工 2020年21期
關鍵詞:催化劑
陳海天,朱峰云,孫云峰,康 艷,王 綱,申玉海,黃太仲
(1.山東魯泰化學有限公司,山東 魚臺 272350;2.山東魯泰控股集團有限公司石墨烯高分子復合材料研發中心 山東 濟寧 272100;3.濟南大學化學化工學院,山東 濟南 250022)
聚氯乙烯(PVC)廣泛應用于國計民生的各個行業,2019年,我國PVC產量約2000萬噸,產值約1500億元,其中超過90%采用“電石法”進行生產,該工藝的最大缺點是采用氯化汞作為乙炔氫氯化反應催化劑,存在嚴重的環境污染。國家發改委發布的《產業結構調整指導目錄》中規定使用高汞催化劑的聚氯乙烯生產工藝和裝置為淘汰產品。2017年,128個國家簽署并開始實施的《水俁公約》約定2031年將關閉原生汞礦的開采。因此,開發無汞乙炔氫氯化催化劑及其相應的生產工藝已成為PVC行業的迫切需求。
作為無汞催化劑的過渡,目前我國PVC行業采取低汞催化劑生產工藝,通過對催化劑載體的選用、預處理與負載工藝優化,提高低汞催化劑的活性和穩定性[1]。唐曉寧[2]和郭燕燕[3]等研究的低汞和無汞催化劑催化乙炔的轉化率可達80%、氯乙烯的選擇性(以下簡稱選擇性)穩定在99%,低汞催化劑并沒有徹底消除行業危機,開發無汞催化劑仍是PVC行業的終極目標。
有研究證明以Au為活性組分、氮改性介孔碳為載體的貴金屬催化劑催化乙炔的轉化率達到89%,選擇性達到99%[4-5]。Zhao Pujun[6]和Bing Wang[7]等發現金屬鈀、銅、銀等金屬的鹽都具有一定的催化活性,其中炔烴配位是催化乙炔氫氯化反應的速率控制步驟。Malta等采用原位X-射線衍射和X-射線吸收精細結構等方法研究了Au催化劑的活性位點[8]。到目前為止,貴金屬催化劑的應用仍然受制于活性低、壽命短、成本高等缺點。
相對于貴金屬催化劑,非貴金屬催化劑顯示出極大的優勢[9],南開大學鄧國才等[10]研究的SnCl2-BiCl3-CuCl2三組分催化劑在140 ℃催化乙炔氫氯化的轉化率和選擇性分別達到97%和95%,但是其使用壽命難于滿足生產需要。清華大學魏飛等[11]開發了以活性炭及SiO2為載體、Bi和P為活性組分的催化劑,結合一定量的助催化劑,其催化乙炔轉化率超過50%,選擇性達到98%,但是反應過程中催化劑表面易于形成積碳導致壽命難以滿足應用要求。魏小波[12]和Di Hu[13]等分別制備了以Bi鹽和其它金屬氯化物、磷酸鹽為活性組分的無汞催化劑,該類催化劑同樣存在催化活性低、壽命短的缺點。
氣固相催化反應過程中傳熱效率低是導致催化劑分解、活性降低的主要原因之一。相對于氣固相反應,液相反應在傳熱方面具有獨特的優勢。日本專利[14]曾報道在液相條件下將氯化金和氯化鉑或氯化鈀負載于活性炭催化乙炔氫氯化反應,其轉化率達60%,選擇性大于99%,壽命約700 h,但是昂貴的價格以及反應中黑色碳化物的產生都限制了其應用。美國專利[15]曾報道在Primene 81-R和Shellsol K混合溶劑中以PdCl2為活性組分的均相催化。相對于傳統的溶液體系,離子液體由于其獨特的物理化學特性也被用于乙炔氫氯化催化反應,據報道離子液體條件下生產氯乙烯的選擇性達到了99%[16-17]。Qin等[18]研究發現[Bmim]Cl溶劑條件下金屬氯化物的催化活性大小順序為:Au≈Pt>Hg≈Cu>Mn>Sn,乙炔最大轉化率為62.5%,選擇性達99%,反應3天后活性基本未變。于志勇[19]等申請了咪唑類離子液體為溶劑,金、鉑、鈀、銠等金屬氯化物為催化劑的專利,實例中列出的催化轉化率介于30%~80%,選擇性大于99%
1 試驗部分
1.1 主要原材料
椰殼活性碳(廣州天金化工有限公司)
AuCl3、NaCl、KCl、CsCl、CuCl2、BaCl2(中國醫藥集團有限公司)
反應氣體:乙炔、氯化氫為自備
1.2 主要儀器設備
熱重-差熱分析儀(TG-DSC)德國Netzsch同步(綜合)熱分析儀 STA
氣相色譜儀 日本島津GC-2014氣相色譜儀
鼓風干燥箱(HTG-9040A)上海精密儀器儀表有限公司
1.3 催化劑制備
設計催化劑的組分中AuCl3和KCl、NaCl和CsCl的比例為1∶9,稱量一定量的催化劑,在高純水中溶解充分混合均勻,然后在110 ℃下進行真空干燥12 h,干燥后的催化劑在進行TG-DSC測試。AuCl3與CuCl2以及NaCl和BaCl2復合催化劑的制備方法與此類似,同樣按一定量的化學計量比稱量個組分后,經溶解后進行共沉淀充分干燥。
椰殼碳經高純水清洗后,鼓風干燥箱130 ℃烘干24 h,然后自然降溫到室溫備用。取一定量椰殼碳,催化劑按照10%的計量比負載在椰殼碳表面,負載催化劑的椰殼碳在130 ℃充分干燥后進行乙炔氫氯化催化性能測試。反應后氣體經氣相色譜測試確定乙炔的轉化率。
2 結果與討論
2.1 催化劑組分設計
氯乙烯合成多相催化研究主要包括進行了非汞催化劑和低汞催化劑研究,重點進行了非汞催化劑的組分、穩定性和催化性能研究。根據已有的研究結果和相關數據,以AuCl3為基本活性組分,設計和優化催化劑的組分,催化劑組分設計見表1。

表1 催化劑的組分設計
2.2 催化劑穩定性測試
根據設計的組分,通過熱重-差熱測試研究催化劑的穩定性,測試氣氛為氮氣,各催化劑在氮氣氣氛的穩定性測試結果如圖1所示。

(a)AuCl3的穩定性測試 (b)AuCl3+KCl的穩定性測試
根據圖1中催化劑AuCl3以及其與KCl、NaCl和CsCl的復合催化劑的熱重-差熱測試結果可以確定各催化劑的起始分解溫度以及最終的分解熱重變化結果,個催化劑的熱分解其實溫度見圖2a,個催化劑的熱重測試結果見圖2b。
由圖2a可見,AuCl3與KCl、NaCl復合后,其分解的起始溫度都有所升高,只有與CsCl復合時分解起始溫度略有降低,但是與此對應的是AuCl3與CsCl復合時催化劑的失重量最小,這應該是由于CsCl中的Cs原子活性較高,其與AuCl3形成了較強的復合作用,從而抑制了AuCl3的分解。由圖2還可以看出,按照NaCl、KCl和CsCl的比例,AuCl3與這些物質相互作用時復合催化劑的分解溫度逐漸降低,但是測試終了時的失重也逐漸降低,這應該歸因于漸進原子外層電子的活性逐漸增加,堿金屬離子與Au離子的作用逐漸增強,從而抑制了AuCl3的分解。
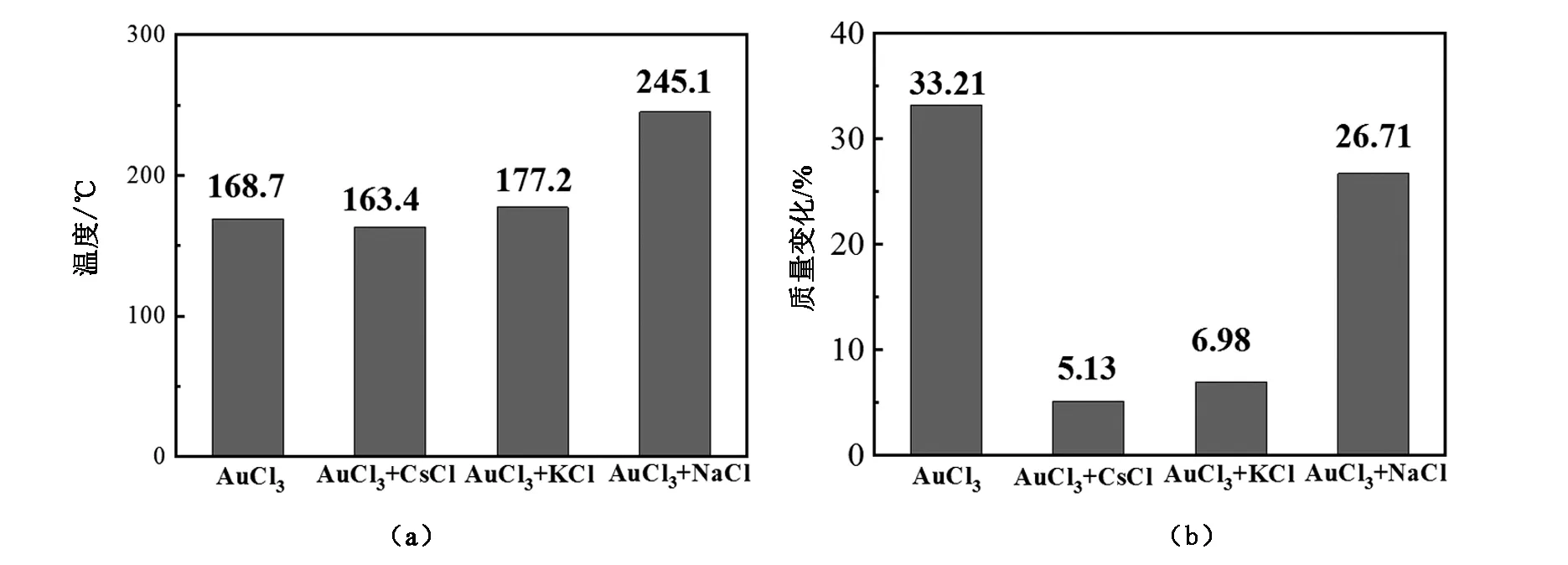
圖2 AuCl3以及其與KCl、NaCl和CsCl的熱分解溫度(a)和熱重分析(b)
AuCl3在與堿金屬進行復合的同時,本論文還研究了AuCl3與CuCl2的復合,并對二者復合催化劑性能進行了熱重-差熱測試,再次基礎上,還研究了AuCl3與CuCl2的復合后與BaCl2和NaCl復合的熱重測試,測試結果見圖3。

(a)AuCl3+CuCl2(1:10)
圖3a可以看出,AuCl3與CuCl2復合時,其初始分解溫度為153.5 ℃,當催化劑與BaCl2進行復合后,其分解溫度僅僅升高到157.5 ℃,這可能是由于堿土金屬Ba離子與Au離子在原子半徑以及電子狀態方面比較接近所造成的。
圖4a表明,當AuCl3與CuCl2和BaCl2復合時,其最大失重僅為2.54%,而與NaCl復合時其失重為2.67%,二者略有差別;但是二者的熱重分解溫度卻有很大的差別,BaCl2復合時催化劑的最高分解溫度為156.5 ℃,而與NaCl進行復合時,其分解溫度為208 ℃,這表明NaCl的復合大大提高了AuCl3與CuCl2的熱穩定性,這應該歸因于Na離子的較小離子半徑以及較強的離子效應。當Na離子引入后,由于Na離子的半徑較小,導致局部電荷密度較大,抑制了Au離子與Ba離子表面的吸附氯離子的分解和脫附,所以導致分解溫度升高到了208 ℃。

圖4 AuCl3與CuCl2以及BaCl2和NaCl的熱失重(a)及分解溫度(b)
2.3 催化劑催化乙炔氫氯化性能測試
催化劑催化乙炔氫氯化測試受催化劑的性能、反應溫度以及空速的影響。根據催化劑的熱重-差熱測試,本文選擇在空速30/h,130 ℃的條件下進行催化劑的氫氯化性能測試,測試反應后乙炔的轉化率以及反應的選擇性。AuCl3、AuCl3+KCl、AuCl3+NaCl、AuCl3+CsCl和AuCl3+CuCl2的催化劑以及以及其AuCl3+CuCl2+BaCl2和AuCl3+CuCl2+NaCl催化乙炔氫氯化測試結果見表2。

表2 各催化劑催化乙炔氫氯化的轉化率和選擇性
由表2可見,AuCl3為基本活性組分的催化劑具有較好的催化乙炔氫氯化性能,其選擇性和轉化率都受到助劑組分以及比例的影響。相對來說,AuCl3+CuCl2為基本組分經BaCl2和NaCl進一步改性的催化劑具有更高的催化性能,這主要應歸因于Ba離子和Na離子的外層電子與Au離子和Cu離子電子的相互作用改善了乙炔和氯化氫在催化劑表面的吸附性能,從而改善了催化劑的性能。
3 結論
本論文的研究結果表明,在椰殼碳表面負載AuCl3為主要活性組分的催化劑具有一定的催化乙炔氫氯化性能,其中堿金屬氯化物與AuCl3的起始分解溫度隨堿金屬離子原子序數的增加逐漸降低,但是與此同時,其催化乙炔氫氯化的活性卻逐漸增強,這可能是由于堿金屬離子外層電子活性增加所導致的堿金屬離子與乙炔和氯化氫的結合性能逐漸增強所導致的。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
