基于深度學習的蛋白質亞細胞定位預測
2020-11-30 05:48:26王藝皓丁洪偉保利勇張穎婕
計算機應用 2020年11期
王藝皓,丁洪偉,李 波,保利勇,張穎婕
(云南大學信息學院,昆明 650500)
(?通信作者電子郵箱893885847@qq.com)
0 引言
隨著蛋白質組學、基因組學等領域研究[1]的快速發展,大量的生物基因信息被不斷挖掘,海量實驗累積的蛋白質數量更是呈指數式增長,傳統的實驗方法耗時費力,已經難以滿足蛋白質定位研究的需要,因此需要通過生物信息學方法進行蛋白質亞細胞定位預測。
近年來,基于機器學習的蛋白質亞細胞定位算法[2-7]已經取得了突破性的進展:Wang 等[2]針對革蘭氏陽性和革蘭氏陰性細菌蛋白提出了兩種有效的多標記預測因子,并通過集成學習的方式進一步優化了分類器性能;Wan 等[3]提出了一種mGOASVM(multi-label protein subcellular localization based on Gene Ontology and Support Vector Machines)算法,該算法將基因本體(Gene Ontology,GO)術語出現頻率引入特征向量的表達,并采用多位點支持向量機(Support Vector Machine,SVM)分類器進行分類預測,最終在Virus proteins和Plant proteins數據集上分別取得了88.9%和87.4%的實際準確率;Wan 等[5]結合了GO 術語出現的頻率與其詞之間的語義相似性,提出了一種HybridGO-Loc(mining Hybrid features on Gene Ontology for predicting subcellular Localization of multi-location proteins)算法,該算法分別在Virus proteins和Plant proteins數據集上取得了93.7%和93.6%的實際準確率。綜上所述,傳統機器學習方法應用于提高蛋白質定位預測的準確性已經取得了相當多的成就,但大多數的傳統機器學習方法仍需通過手工操作來表示特征,而深度學習的出現良好地解決了這個問題。
與傳統的機器學習方法相比,深度學習能夠通過多層次深度網絡結構從輸入數據中自動學習良好的特征表示。經典的深度學習框架主要有深度置信網絡(Deep Belief Network,DBN)[8]、堆棧式自編碼器(Stacked AutoEncoder,SAE)[9]和卷積神經網絡(Convolutional Neural Network,CNN)[10]等。由于深度網絡強大的學習能力和泛化能力,近幾年已經開始逐漸應用于生物信息學領域[11-14]中。例如:Wen 等[11]利用DBN 提出了一種用于預測藥物-靶標之間的相互作用的深度學習算法——DeepDTIs(Deep learning-based Drug-Target Interaction prediction),該算法性能超過了當時最先進的傳統機器學習算法;Alipanahi等[12]基于CNN 提出了一種稱為DeepBind的深度學習算法,該算法用于預測DNA和RNA結合蛋白的序列特異性,取得了不錯的效果;Liu 等[13]結合支持向量機和深度神經網絡提出了一種用于蛋白質折疊識別的算法——DeepSVMfold,該算法取得了明顯優于其他傳統機器學習算法的性能表現。隨著越來越多的研究者們將注意力轉移到深度學習的應用研究中,大量新穎有效的深度學習算法不斷涌現,這也為蛋白質亞細胞的定位預測研究工作提供了一定的便利條件。
張穎婕[7]基于特征融合和集成學習的思想,結合了偽氨基酸組成法(Pseudo-Amino Acid Composition,PseAAC)、偽位置特異性得分矩陣(Pseudo Position Specific Scoring Matrix,PsePSSM)和三肽組成三種特征提取方式,然后通過主成分分析法(Principal Components Analysis,PCA)降維,最后輸入集成SVM 分類器完成了蛋白質定位預測任務。雖然文獻[7]取得了較好的預測準確率,但是也帶來了以下幾個問題。
首先,采用PCA 對融合后的特征向量進行降維處理,雖然可以有效剔除冗余信息并避免維度災難帶來的影響,但同時它也會帶來一些消極影響:1)對于主成分的解釋往往具有一定的模糊性,進而導致降維后的特征表示可能與原始數據有所差異;2)某些主成分雖然貢獻率小,但是它們往往包含了關于樣本差異的重要信息,特別是像蛋白質這樣的不平衡數據集;3)PCA作為無監督學習算法,仍需通過手工操作來確定主成分和表示特征。
其次,集成支持向量機的使用,雖然在一定程度上提高了預測準確率,但同時它也增加了算法復雜度。
本文針對以上問題,對特征提取模型和分類器進行了改進和優化。首先對PseAAC 和三聯體編碼法(Conjoint Traid,CT)進行了改進,進一步豐富了特征融合后的蛋白質序列表征模型;接著將融合后的特征向量輸入到本文構造好的堆棧式降噪自編碼器(Stacked Denoising AutoEncoder,SDAE)深度網絡,SDAE 網絡可以進一步深入學習到表達能力更強、泛化能力更好、更接近真實數據的特征表示,避免了PCA 降維對蛋白質序列表征模型產生的消極影響;然后輸入Softmax 回歸分類器進行分類預測,這降低了算法的復雜度;最后采用留一法分別在Virus proteins 和Plant proteins 數據集上進行交叉驗證,并將實驗結果與其他現有算法進行比較。實驗結果表明,本文提出的新方法能夠有效提高蛋白質亞細胞定位預測的準確性。
1 構建多位點蛋白質序列特征提取模型
1.1 改進型偽氨基酸組成模型
Chou[15]提出了偽氨基酸組分(PseAAC)方法,在氨基酸組分法(Amino Acid Composition,AAC)的基礎上引入了λ 階相關因子更好地表達序列信息。傳統的PseAAC 模型僅考慮了疏水性、親水性和側鏈分子量三種理化特征,本文在此基礎上增加了極性、極化率、溶劑化自由能、曲線形狀指數、轉移自由能、氨基酸組分、回歸分析相關系數、殘基可及表面、分配系數、氨基酸邊鏈體積、表面區域溶解能力、網絡負荷指數共12種氨基酸理化性質,構造了一種包含15 種氨基酸理化性質的改進型PseAAC模型。
根據改進型PseAAC模型,每條蛋白質序列可以表示為:

其中每個元素pu可由式(2)求出:
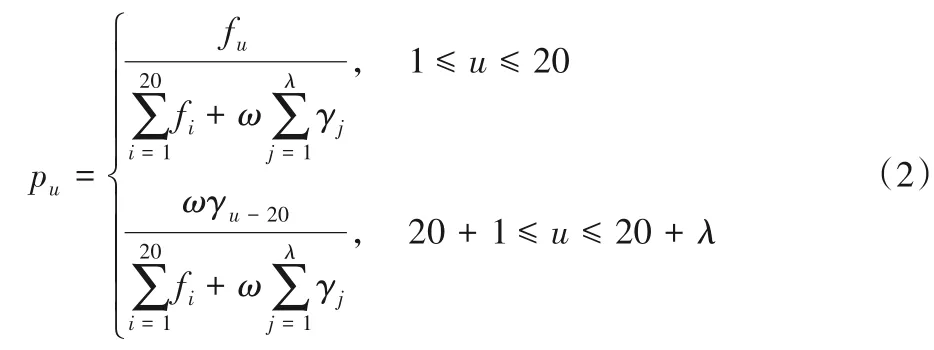
其中:fu表示每種氨基酸在蛋白質序列中出現的概率;ω 是權重因子,本文默認取0.05;γj表示j個緊鄰相關因子,反映了不同氨基酸之間的順序信息,可由式(3)求得:

Ji,i+k稱為相關函數,其定義為:
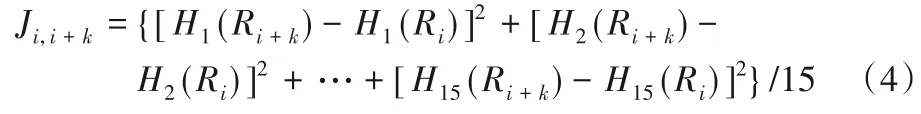
其中H(Ri)由式(5)求得:

其中:h0(Ri)為氨基酸Ri對應理化性質的原始特征值為對應理化性質下20種氨基酸原始特征值的平均值,ν(h0)表示其對應的方差。
由于λ 的取值會影響最終分類預測的結果,故要通過實驗選取最佳參數。實驗中λ 分別取1~30,輸入支持向量機在數據集上進行實驗,采用留一法對預測結果進行檢驗,通過比較得出,當λ=15時預測準確率達到最高。因此,通過改進型PseAAC,一條蛋白質序列可以轉化為一個35維的特征向量。
1.2 偽位置特異性得分矩陣
Jones[16]提出了位置特異性得分矩陣(Position Specific Scoring Matrix,PSSM),該方法充分考慮了氨基酸的序列進化信息。本文選用了PSI-BLAST[17]來獲取PSSM 矩陣,設置閾值為0.001,最大迭代次數為3,選取NCBI 的非冗余蛋白質數據庫(non-redundant,nr)[18]作為對比,其下載網址為ftp://ftp.ncbi.nih.gov/blast/db/nr。由此可以獲得一個L × 20 的PSSM矩陣,即:


由于不同蛋白質序列的長度L 是不同的,故需要將不同蛋白質序列的PSSM 矩陣轉化為維度相同的矩陣。從PSSM矩陣中提取氨基酸組分(AAC)則得到了PSSM-AAC模型,即:
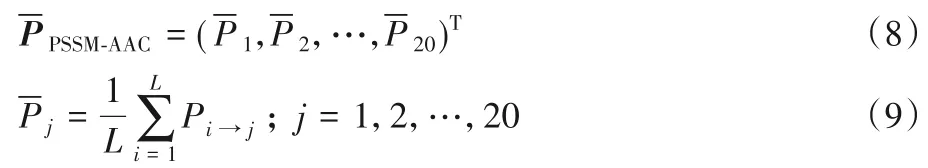

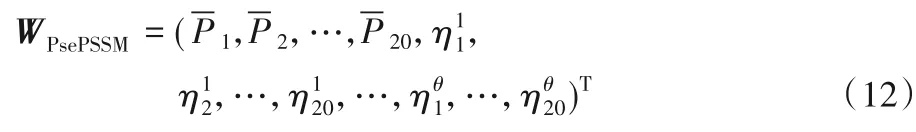
1.3 三聯體編碼
Shen 等[20]提出了三聯體編碼方式(CT)用于預測蛋白質相互作用的工作中,該方法考慮了蛋白質序列中相鄰氨基酸分子之間的相互作用。氨基酸的分類依據決定了三聯體的構成方式,與按照親疏水性劃分為6 類[7]不同,本文根據偶極性和側鏈體積將20 種氨基酸重新劃分成7 類,接著再引入緊鄰三聯體,將連續的三個氨基酸看作是一種三聯體結構,故可得三聯體共有343(7×7×7)種構成方式。由CT 可得,每條蛋白質序列有343 個特征因子fi。由于fi的大小與蛋白質序列的長度成正比關系,而且不同的蛋白質序列長度相差較大,故要進行歸一化處理,引入以下定義:
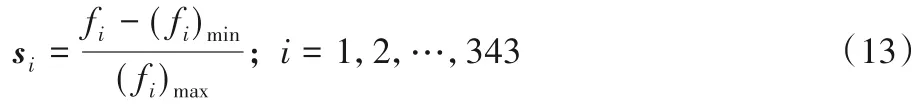
其中:si表示標準化后的特征向量,且si∈[0,1]。接下來將蛋白質對的兩條序列串聯起來以表示其相互作用信息:

其中DA和DB表示蛋白質對的兩條序列。綜上可得,每條蛋白質序列可以轉為343(7×7×7)維的特征向量表示,即:
在企業績效管理工作的開展中,最終目的都是將企業績效管理工作的開展提升整體企業業績開展能力,并且在企業業績開展能力的提升過程中,能夠將對應的績效管理工作和對應的績效管理因素協調好。保障了績效管理因素控制關系的協調性建設,同時按照這種績效管理因素的考核實施來看,在石油裝備企業的建設和管理中,要想保障HU績效考核管理體系建設能夠滿足石油裝備企業的自身性績效管理工作開展需求,對應的石油裝備企業績效考核管理者,應該在績效考核管理工作的開展中,將對應的績效考核管理工作與激勵制度的建設結合在一起,這樣不僅能夠調動企業員工的工作積極性,同時也能夠提升績效考核管理效率,保障了企業的科學化績效管理。

1.4 多特征融合
本文基于多特征融合的思想,將改進型PseAAC、PsePSSM 和三聯體編碼法三種特征提取方式結合,構成了一種全新的蛋白質序列特征提取模型。融合之后的蛋白質序列信息可由式(16)表達:
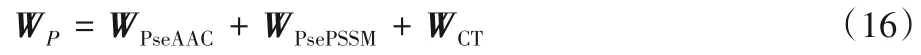
將數值代入之后,每條蛋白質序列可以轉化為458(35+80+343)維特征向量表示。
2 堆棧式降噪自編碼器
堆棧式降噪自編碼器(SDAE)[21]是由多個降噪自編碼器(Denoising AutoEncoder,DAE)逐層連接而成的一種深度神經網絡結構。它通常主要包括兩個過程:無監督的預訓練和有監督的微調。預訓練過程會以無監督方式逐層學習深層特征并初始化深度網絡的參數,同時它會使用反向傳播算法以微調的監督方式進一步優化預訓練過程生成的參數,從而提升模型性能。因此,SDAE 具有良好的學習能力和泛化能力。SDAE模型架構如圖1所示。
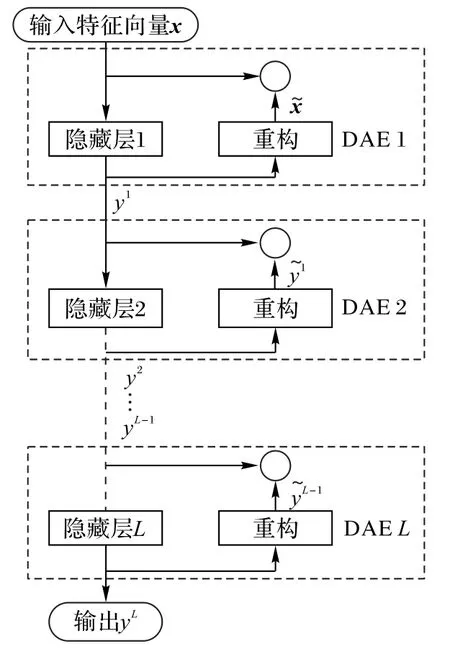
圖1 SDAE模型架構Fig.1 Model architecture of SDAE
自編碼器(Auto Encoder,AE)是一種無監督表征學習的高效深度神經網絡,本文默認輸出層與輸入層參數相同。AE由編碼器和解碼器組成。假設AE輸入特征向量其中dx表示輸入的維數,編碼器通過以下映射函數h將x從輸入層投影到隱藏層y ∈Rdy:

其中:W 表示dy× dx權重矩陣表示映射到隱藏層對應的向量維度)表示偏差向量。本文選取ReLU(Rectified Linear Unit)函數作為激活函數af。
在解碼器中,隱藏層y 通過以下映射函數h*映射到輸出層

在AE 中,每個輸入特征向量xi通過函數h 映射到隱藏層yi,再通過函數h*映射到輸出層輸出重構向量。為了使重構輸出與輸入x 盡可能相似,本文選取ReLU 函數作為隱藏層的激活函數af,選取Softplus 函數作為重構層的激活函數,同時引入以下均方誤差:
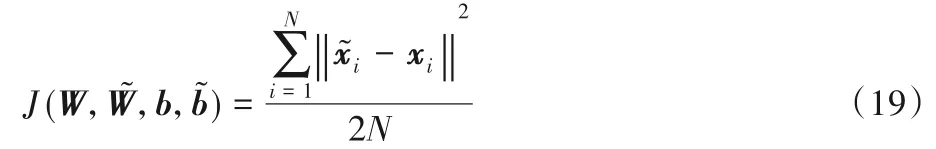
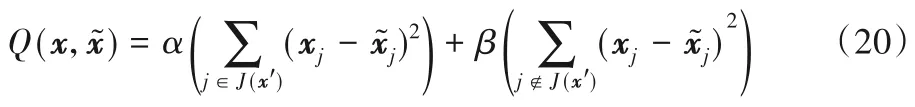
其中:α 為噪聲污染維度重構代價權重,β 為無噪聲污染維度重構權重。
本文在此基礎上,提出了一種堆棧式降噪自編碼器(SDAE)用在蛋白質定位預測任務中。SDAE 訓練過程如圖2所示。
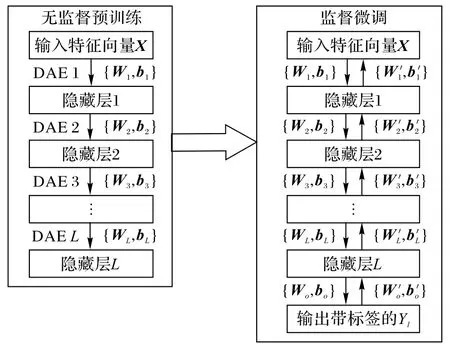
圖2 SDAE訓練流程Fig.2 Training flow chart of SDAE
如圖2 所示,SDAE 主要進行兩個步驟,分別是無監督預訓練和監督微調。在預訓練中,對于首個DAE 按照最小化重構誤差原則將輸入的原始特征向量映射到第一個隱藏層,訓練完首個DAE后得到參數{W1,b1},接著將第一個隱藏層的輸出會作為第二個隱藏層的輸入,繼續訓練第二個DAE 得到參數{W2,b2}。以這樣的方式來對整個SDAE 層進行逐層預訓練,直到得到最后一個DAE 層。在無監督預訓練之后,通過預訓練得到的權重參數{Wk,bk}(k=1,2,…,K)來初始化每個隱藏層的權重,然后通過反向傳播對整個深度網絡進行微調,通過最小化目標變量的預測誤差來獲得更新權重,k=1,2,…,K。其反向傳播函數定義如下:

其中:rj表示第j條蛋白質序列標記值表示其預測值。SDAE深度網絡最后一層選用Softmax 回歸函數進行分類。本文SDAE算法采用DeepLearing Tutorials 軟件包在Matlab2018a中實現,實驗環境為Intel Core i7-9750H CPU 2.90 GHz 16.0 GB。
3 性能評估
目前,常用于模型性能檢測的主要方法有獨立性檢驗、自相容檢驗、K 折交叉驗證和留一法(leave-one-out cross validation)等。其中,留一法由于其客觀公正的特點,被廣泛應用于蛋白質亞細胞定位預測模型的性能評估工作當中[23]。故本文選取留一法對模型性能進行評估。對于評估指標,本文采用生物信息學中最常用的5個指標[24]來對模型性能進行全方位的評估:

4 實驗結果與分析
4.1 數據集
本文選用數據集來自被研究者們廣泛認可和使用的Plant proteins、Viral proteins 兩個數據集(可以從http://www.csbio.sjtu.edu.cn/bioinf/下載):數據集Plant proteins 共包括1 055 條蛋白質序列,涉及12 個亞細胞位點標簽;數據集Viral proteins 共包含252 條蛋白質序列,涉及6 個亞細胞位點標簽。數據集的詳細信息如表1所示。
在Plant proteins 和Viral proteins 兩個數據集中,同時擁有大量的單位點蛋白和多位點蛋白,適用于本文對于蛋白質亞細胞的多標簽分類預測的研究。其具體位點分布情況如表2所示。

表1 實驗中使用的Viral proteins和Plant proteins數據集Tab.1 Viral proteins and Plant proteins datasets used in the experiment

表2 Viral proteins和Plant proteins數據集中蛋白質序列位點分布情況Tab.2 Distribution of protein sequence sites in Viral proteins and Plant proteins datasets
4.2 實驗結果與分析
4.2.1 特征提取算法性能分析
首先在Viral proteins 和Plant proteins 數據集上分別用改進型PseAAC、PsePSSM、三聯體編碼法、多特征融合法和本文提出的新方法進行實驗并使用留一法進行驗證,其實驗結果如表3所示。
從表3可以看出,多特征融合法由于結合了前三種特征提取方法構造了更為豐富的蛋白質序列表征模型,其分類預測結果在各項指標上完全碾壓其他三種單一特征提取方法。多特征融合法與三種單一特征提取方法中表現最好的改進型PseAAC 相比,其Coverage、Aiming、Accuracy 和Absolute True均提升了6個百分點以上,同時Absolute False降低了1個百分點左右。同時可以看出,本文方法由于加入了堆棧式降噪自編碼深度(SDAE)網絡進一步篩選并提取了更加魯棒和真實的特征表示,其實驗各項指標相對多特征融合法來講又有了大幅度的提升。對于Viral proteins數據集,本文方法和多特征融合法相比,前者的Coverage、Aiming、Accuracy 和Absolute True 分別比后者高出了4.1、4.91、0.94 和1.6 個百分點,而Absolute False 降低了0.58 個百分點;對于Plant proteins 數據集,本文方法的Coverage、Aiming、Accuracy 和Absolute True 分別比多特征融合法提高了4.25、5.75、1.51和3.93個百分點,而Absolute False 降低了1.27個百分點。綜上所述,本文方法可以有效提高多位點亞細胞分類預測的準確性。

表3 不同方法在Viral proteins數據集和Plant proteins數據集上的實驗結果對比 單位:%Tab.3 Comparison of experimental results of different methods on Viral proteins dataset and Plant proteins dataset unit:%
4.2.2 分類器性能分析
本節主要對目前用于多位點亞細胞定位任務四種表現較好的分類器進行了實驗和對比。這四種分類器分別為:樸素貝葉斯(Naive Bayesian,NB)、SVM、隨機森林(Random Forests,RF)以及Softmax 回歸。首先將SDAE 網絡得到的特征向量分別輸入NB、SVM、RF 和Softmax 回歸分類器中,采用留一法在Viral proteins 和Plant proteins 兩個數據集上進行交叉驗證。其中,NB和RF均采用默認參數;SVM 中的核函數選擇高斯核函數。實驗結果如圖3所示。
由圖3 可以看出,在Viral proteins 數據集上,NB、SVM、RF分別取得了91.2%、93.7%和96%的整體準確率,而本文所選用的Softmax回歸分類器取得了98.2%的整體準確率,相比前三種分類算法分別提高了7、4.5 和2.2 個百分點;而在Plant proteins 數據集上,NB、SVM、RF 分別取得了89.5%、92.9%和95.2%的整體準確率,本文所選用的Softmax 回歸分類器取得了97.6%的整體準確率,相比前三種分類算法分別提高了8.1、4.7 和2.4 個百分點。綜上所述,本文所選用的Softmax回歸分類器分類效果最好。
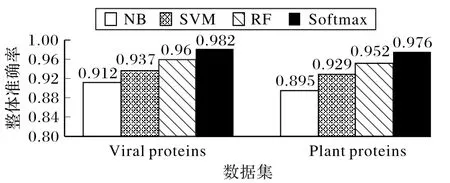
圖3 在兩個數據集上四種分類算法的預測結果對比Fig.3 Comparison of prediction results of four classification algorithms on two datasets
4.2.3 與其他算法比較
接下來將本文方法所取得的實驗結果與其他現有算法模型取得的實驗結果進行對比,均采用留一法進行測試。先依次對各位點標簽所取得的預測結果進行分析比較,在Viral proteins數據集上的實驗對比結果如表4所示。
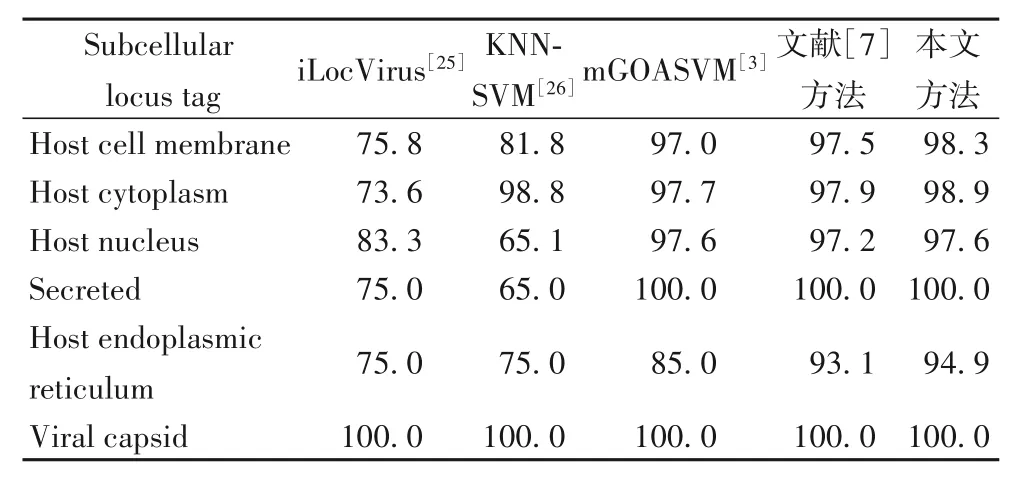
表4 Viral proteins數據集上不同方法性能的比較結果單位:%Tab.4 Performance comparison of different methods on Viral proteins dataset unit:%
由表4 可以看出,本文所提新方法與iLoc-Virus、KNNSVM、mGOASVM 算法的實驗結果相比,均有較明顯的提高。特別是與文獻[7]算法相比,本文方法在Host cell membrane、Host cytoplasm、Host nucleus 和Host endoplasmic reticulum 位點上的預測準確率均有不同程度的提升,說明了本文在特征融合后引入SDAE 深度網絡的有效性和科學性。為了進一步驗證新方法的優越性,將預測結果與現有算法中表現較好的mGOASVM 和文獻[7]算法進一步進行比較,其詳細對比結果如表5所示。
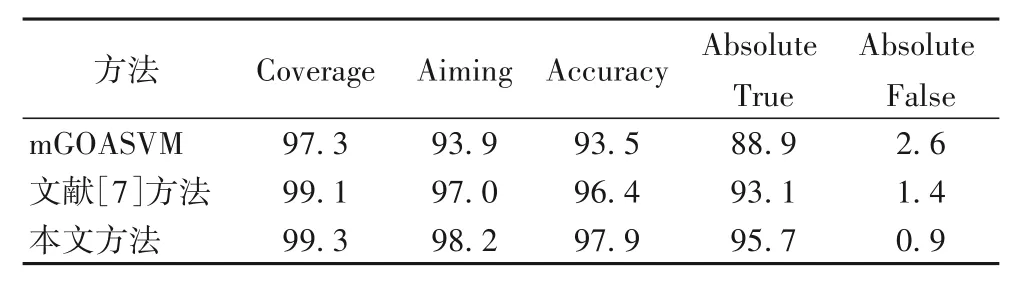
表5 Viral proteins數據集上三種方法的實驗性能對比 單位:%Tab.5 Comparison of the experimental performance of three methods on Viral proteins dataset unit:%
由表5 可得,在Viral proteins 數據集上,本文方法與mGOASVM 算法相比,前者的Coverage、Aiming、Accuracy 和Absolute True 分別比后者提高了2、4.3、4.4 和6.8 個百分點,其Absolute False 降低了1.7 個百分點,可以看出整體提升幅度還是蠻大的;進一步分析,本文方法與文獻[7]相比,其Coverage、Aiming、Accuracy 和Absolute True 分別提升了0.2、1.2、1.5 和2.6 個百分點,而Absolute False 降低了0.5 個百分點,可以發現整體提升效果還是很明顯的。綜上可得,本文方法在數據集Viral proteins上表現出了良好的分類預測性能。
為了進一步驗證本文方法的優越性,繼續在數據集Plant proteins 上進行實驗分析,將各位點標簽上得到的預測結果與其他現有算法模型取得的實驗結果進行比較,其對比結果如表6 所示。由表6 可知,與傳統的蛋白質定位算法iLoc-Plant相比,本文方法在蛋白質各位點標簽上的預測準確率有顯著的提升。而相較于mGOASVM 和HybridGO-Loc 這兩種算法,本文方法除了在Cell wall proteins 和Mitochondrion proteins 位點的預測準確率稍有下降以外,其他位點的預測準確率均有一定程度的提高;與文獻[7]算法相比,本文方法除了在Nucleus proteins 位點預測準確率稍有下降,其他位點的預測準確率都基本提升和持平。特別的,本文方法在Extracell proteins、Peroxisome proteins、Plastid proteins 和 Vacuole proteins 位點上取得了100%的預測準確率。由于這四種方法各位點亞細胞預測準確率較為接近,為了進一步驗證本文方法的有效性,接下來引入多標簽預測評估指標,對四種方法進一步分析比較,其詳細對比結果如表7所示。
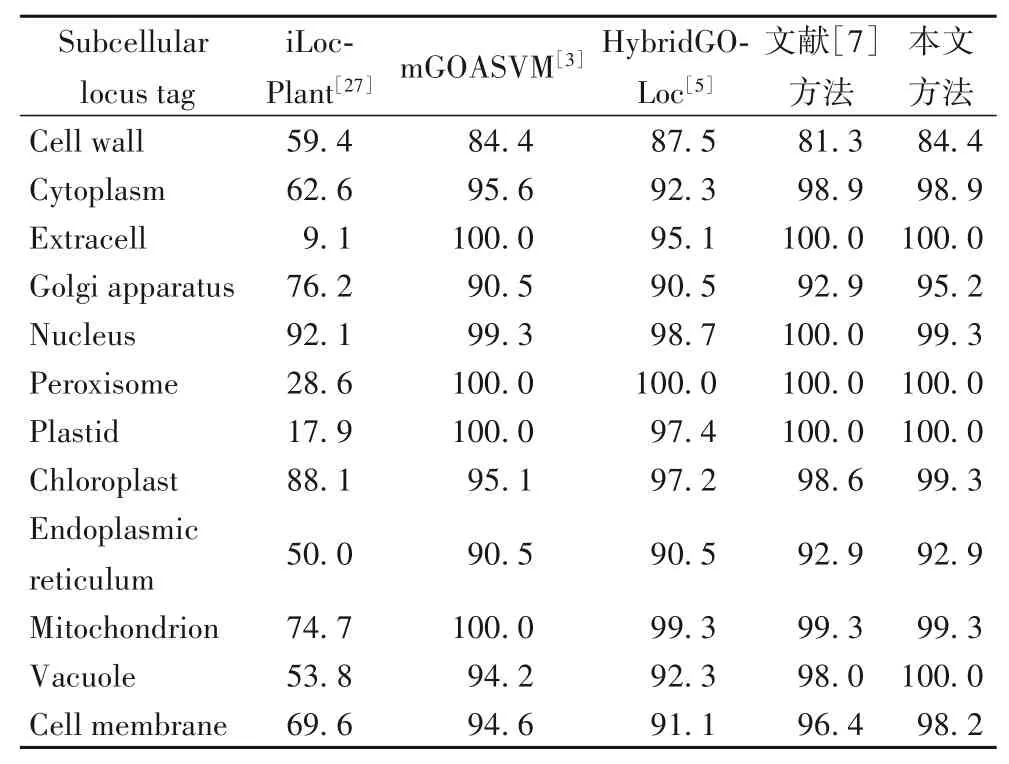
表6 Plant proteins數據集上不同方法性能的比較結果 單位:%Tab.6 Performance comparison of different methods on Plant proteins dataset unit:%
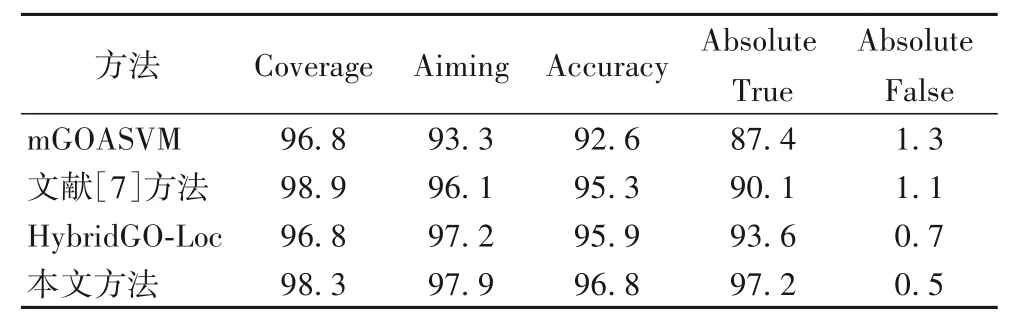
表7 Plant proteins數據集上四種方法的實驗性能對比 單位:%Tab.7 Comparison of the experimental performance of four methods on Plant proteins dataset unit:%
由表7 可知,在Plant proteins 數據集上,HybridGO-Loc 算法的分類預測效果明顯要優于mGOASVM 和文獻[7]算法,故本文方法與三種算法中相對表現更好的HybridGO-Loc 算法相比,其Coverage、Aiming、Accuracy 和Absolute True 分別提升了1.5、0.7、0.9 和3.6 個百分點,而Absolute False 降低了0.2個百分點。進一步分析,本文方法與文獻[7]方法相比,雖然其Coverage 略有下降,但其他指標均有明顯改善,其Aiming、Accuracy 和Absolute True 分別提升了1.8、1.5 和7.1 個百分點,而Absolute False 降低了0.6個百分點,這再一次證明了本文方法優化策略的有效性和科學性。綜上所述,本文方法能有效提高多位點亞細胞定位的預測效果。
5 結語
本文提出了一種基于深度學習的蛋白質亞細胞定位預測新方法。首先,分別通過改進型PseAAC、PsePSSM 和三聯體編碼法對蛋白質序列信息進行特征提取,并將三種方法提取的特征向量進行融合,構造了一種全新的蛋白質序列信息表達模型,該模型不僅包含了蛋白質序列中氨基酸的理化性質、頻率信息和順序信息,還充分考慮了氨基酸之間的進化信息以及相互作用,進一步豐富了蛋白質序列表達信息;接著,將融合后的特征向量輸入SDAE 深度網絡,通過預訓練和微調的方式得到最優的深度學習網絡,該網絡可以自動學習并提取更加魯棒、真實的特征表示信息;然后,輸入Softmax 回歸分類器進行分類預測;最后,采用留一法在Virus proteins 和Plant proteins 數據集上進行交叉驗證。通過將實驗結果與多種現有算法進行比較,充分證明了新方法可以有效提高蛋白質亞細胞定位預測的準確性。下一步將繼續擴大數據集,在此基礎上豐富蛋白質序列表征模型,并對深度學習網絡進行優化,進一步提高蛋白質亞細胞定位預測的準確性。
猜你喜歡
中學生數理化·七年級數學人教版(2020年11期)2020-12-14 06:59:52
兒童故事畫報(2019年5期)2019-05-26 14:26:14
藝術品鑒證.中國藝術金融(2018年8期)2019-01-14 01:14:28
藝術品鑒證.中國藝術金融(2018年10期)2019-01-08 02:44:26
藝術品鑒證.中國藝術金融(2018年6期)2019-01-08 02:43:04
藝術品鑒證.中國藝術金融(2018年12期)2018-08-26 06:03:48
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
新聞傳播(2015年10期)2015-07-18 11:05:40
