淺析電解質中離子輸運的微觀物理圖像*
2020-12-05 07:34:02任元鄒喆乂趙倩王達喻嘉施思齊
物理學報 2020年22期
任元 鄒喆乂 趙倩 王達 喻嘉 施思齊?
1) (上海大學材料科學與工程學院, 上海 200444)
2) (內蒙古科技大學機械工程學院, 包頭 014010)
3) (湘潭大學材料科學與工程學院,湘潭 411105)
4) (上海大學材料基因組工程研究院, 上海 200444)
解析離子在電解質中的輸運特征所表現出的微觀物理圖像, 對于調控離子傳導行為具有重要的指導意義. 本文系統總結了離子在液態、有機聚合物和無機固態電解質中的離子輸運物理圖像及其影響因素, 通過分析各種輸運物理模型并比較三類電解質中的離子輸運機制, 提煉出勾勒離子輸運物理圖像的相關描述因子. 輸運介質的物理形態從連續流體到柔性載體再到剛性骨架的演變過程中, 離子輸運圖像由各類電解質的固有屬性與外部條件共同刻畫, 其中介質無序性占據主導作用. 揭示電解質結構和離子電導率及輸運過程等動力學行為之間的科學規律, 有利于發展基于離子輸運微觀物理圖像的傳導離子動力學性能調控方法.
1 引 言
擴散指物質通過原子或分子的熱運動進行位置遷移, 其速度在氣體、液體、固體中依次變慢. 擴散在許多科學與工程問題中起著關鍵作用, 例如:離子輸運、中子傳輸、反應物聚集等的速度均受擴散限制[1]. 在研究中通常“擴散”是指一種宏觀的物理現象, “輸運”是離子主動或被動地在介質中運動的特性, “遷移”被用來描述離子的具體運動形式,離子在介質中的運動過程常被描述為“傳導”. 對擴散深入了解, 需獲取有關離子位置及其在介質中移動方式等信息. 準確地利用離子擴散的信息可以獲得快速的擴散路徑, 進而提高介質的離子電導率.由離子擴散信息所描述的輸運圖像對于物質傳輸和離子傳導至關重要[1].
作為電池體系得以正常工作的關鍵步驟之一,離子輸運的快慢對其循環性能、充放電速率等指標具有重要影響[2,3]. 為滿足高能量密度電池體系的需求, 各類新型電極材料被提出, 設計與之相適應的電解質迫在眉睫. 顯然, 由輸運圖像決定離子電導率的電解質對電池性能起著關鍵作用[3-7]. 為獲取刻畫電解質中離子輸運微觀物理圖像的描述因子, 可從電解質的宏觀物理形態入手劃分為獨立又交叉的三類: 液態電解質、聚合物基電解質、無機固態電解質. 圖1 總結了可供鋰離子或鈉離子輸運的各類電解質: 從基于連續流體的液態電解質逐漸過渡為柔性載體性質的聚合物基電解質, 再演化到剛性骨架的無機固態電解質. 盡管物理形態是一種連續的演化過程, 但該過程中離子輸運的微觀圖像表達卻各具特征[8,9].
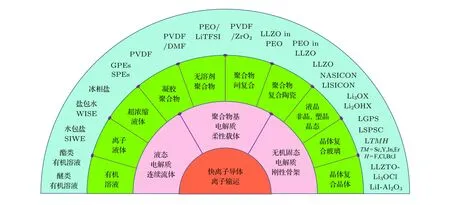
圖1 可供離子輸運的電解質, 包括: 液態電解質、聚合物基固態電解質、無機固態電解質以及復合固態電解質Fig. 1. The electrolytes for ion transport: liquid electrolyte, polymer-based solid electrolyte, inorganic solid electrolyte and composite solid electrolyte.
電解質由液態到固態的演變中, 跨電解液的離子傳輸方式可以整體上描述為介質與結構方式兩種(vehicular versus structural ion motions)[10].常規液態電解質溶劑化離子與溶劑化鞘同時輸運,而固態電解質則是溶劑化位點被固定, 輸運離子則通過耦合-解離過程或者頻繁交換可移動的溶劑分子躍遷, 這是液態與固態條件的兩種極端輸運形式. 一般情況下兩類離子輸運方式可以并行, 隨著電解質濃度的變化, 離子輸運逐漸由介質主導轉變為結構主導, 極端實例即為擁有高機械強度的固態電解質[10]. 固態電解質作為電池固態化的關鍵技術, 相應的離子輸運特性以及界面問題尤為關鍵[2].無論是柔性聚合物電解質還是無機固態電解質, 與正負極接觸界面離子輸運均為研究焦點, 其中固-固點接觸會阻礙離子傳輸路徑, 由此變化的離子輸運微觀圖像對全固態電池體系的性能影響更大. 因此, 為保證較高的離子電導率與避免界面問題, 可以設計基于兩種或多種無機電解質的復合電解質,可提供多種離子輸運機制[8,11].
從具體的離子輸運動力學過程角度看, 液態電解質離子輸運過程中離子的溶劑化與去溶劑化是主導過程, 聚合物固態電解質主要以配位傳遞輸運離子. 當介質趨于骨架化后離子則趨于獨立化運動, 且輸運離子與介質間的相互作用也會發生變化. 骨架達到一定剛度, 無機固體電解質中的離子輸運開始趨于通道化運動模式, 其中輸運離子與骨架離子間相互作用成為離子輸運圖像的重要因子.因此在無機固體電解質中, 離子間相互作用帶來的離子遷移各向異性對離子輸運動力學有重要影響.
2 離子輸運機制
2.1 離子輸運理論計算模型
離子輸運特性的表現形式, 通常以離子電導率、離子遷移數、擴散系數以及離子遷移擴散能壘等物理量組成圖像描述因子. 連續性流體、柔性聚合物載體以及剛性骨架電解質的離子電導率的計算, 通常基于Fick 定律(Fick 第一定律), 即通過搭建鹽在水中擴散的理論模型, 建立鹽擴散流量與其在水中的濃度梯度間正比關系, 并定義為擴散系數. 擴散系數包括: 自擴散系數、缺陷擴散系數、化學擴散系數等. 自擴散系數表示離子布朗運動的活躍性, 采用同位素測定法可獲得同位素擴散系數,但通常略小于離子自擴散系數; 缺陷擴散系數由缺陷躍遷活化能決定, 描述自擴散系數的活化能由缺陷形成能和缺陷躍遷能組成; 化學擴散系數用于描述具有可支配擴散的固相反應速率, 例如Li+嵌入TiS2中的擴散現象. 為描述離子擴散連續性位移函數隨時間變化的動態過程, Fick 擴散方程(Fick 第二定律)進一步被提出. 在較復雜的輸運體系中, 離子擴散濃度隨時間變化值與其濃度梯度的一次微分的比例系數被定義為互擴散系數, 其影響因素較多, 包括溫度、壓力、組分間相互作用等.Fick 定律由(1)式表述[12-14]:
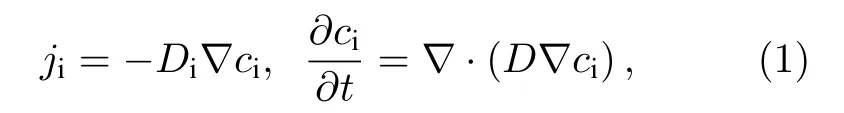
其中j為離子擴散流量(cm2·s);Di為離子擴散系數(cm2/s);ci為離子擴散濃度(離子/cm3);為濃度梯度.
對于玻璃化轉變溫度以上的離子液體電解質、有機溶液電解質、凝膠聚合物電解質、聚合物固態電解質等, 離子輸運行為通常采用Vogel Tamman Fulcher (VTF)理論模型(2)表述[15]:
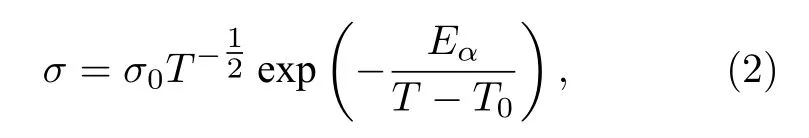
其中σ0為與載流子數量相關的頻率因子;Eα為離子輸運的活化能;T為工作溫度;T0為平衡玻璃轉移溫度(T0=Tg— 50,Tg為玻璃化轉變溫度).
對于玻璃轉化溫度以下的非晶聚合物電解質,離子輸運行為采用Arrhenius 模型(3)表述[16]:
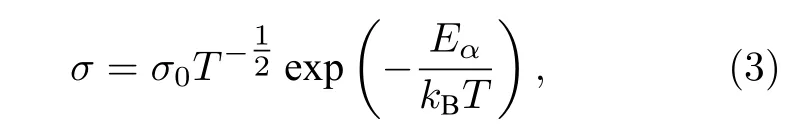
其中k為玻爾茲曼常數, 其余參數與(2)式中表述一致.
無機固態電解質中離子輸運通常基于離子躍遷模型描述, 相關的結構因子與離子電導率可以用與離子導體基本躍遷相關的參數表示. 結構描述因子通常受到晶格體積、離子輸運瓶頸尺寸、配位、骨架無序、骨架畸變、缺陷以及晶格動力學等因素的影響, 同時因子之間也存在相互制約關系. 離子躍遷模型由(4)式表述[1,17]:
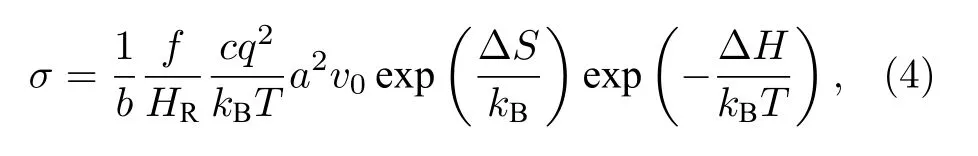
其中b為幾何結構因子(一維遷移b= 2, 二維遷移b= 4, 三維遷移b= 6);c為輸運離子濃度;q為輸運離子電荷;a為跳躍距離;ν0為嘗試跳躍頻率;T為絕對溫度;f為相關因子, 通過示蹤擴散系數與隨機擴散系數比值描述單離子自相關性[1,9,18];HR為Haven 比, 通過離子導體中長程擴散系數與示蹤擴散系數比值描述多離子互相關性[9,19]; ΔS為遷移熵, ΔH為活化能, 均對離子電導率有影響[20]. 擴散方程的建立對于通過理論計算方法研究離子輸運至關重要, 并被廣泛應用.2012 年Shi[21]構建了基于Fick 定律的多離子協同輸運擴散方程, 并應用于Li2CO3擴散機制的研究中.
如圖2 所示, 離子輸運的微觀物理圖像的刻畫受到遷移路徑、擴散能壘、過渡態以及運動方式四種關鍵因素的交叉影響, 同時各成像描述因子進一步衍生出更為具體的諸多勾勒元素. 實現快離子傳導的必要條件包括: 1)離子輸運遷移路徑多樣化,通過路徑從平面遷移到空間擴散以實現各向異性;2)通過骨架無序、瓶頸尺寸以及勢能梯度的調節使得擴散能壘趨于降低; 3)依靠骨架剛度與擴散鞍點數量的調控減少離子輸運過渡態的數量; 4)離子運動方式的多樣化所誘導的不同輸運機制對物理圖像的影響更為重要. 高層次的離子輸運圖像描述因子之間互相牽制, 底層的勾勒元素之間相互影響, 以及描述因子與勾勒元素之間跨級交叉作用,共同刻畫出離子輸運的微觀物理圖像[20]. 除電解質本質屬性的影響, 電解質在電池體系中所處環境對離子輸運的影響不容忽視. 例如: 電解質與正負極間的電位不同, 導致載流子濃度不同, 進而影響離子輸運圖像[21].
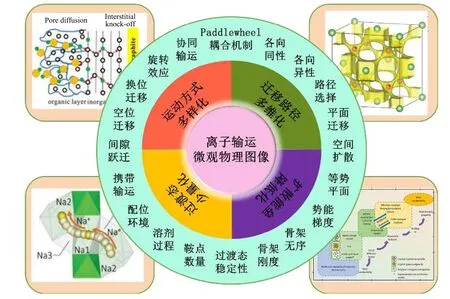
圖2 影響離子輸運微觀物理圖像的因素包括輸運機制與描述因子. 如: 左上角knock-off 離子輸運[21]; 右上角: BVSE 方法描述離子輸運通道[22]; 左下角: NASICON 中多離子協同輸運[23]; 復合結構電解質離子輸運[8]Fig. 2. The factors affecting the microscopic physical image of ion transport: transport mechanism and description factors. For example: knock-off ion transport[21], BVSE method based ion transport channel description[22], multi-ion coordinated transport in NASICON[23], mobile ion in composite solids[8].
2.2 電解質中離子輸運機制
離子在電解質中的輸運圖像描述因子與傳導介質的物理屬性直接相關. 從屬于連續流體的液態電解質, 到擁有柔性載體的聚合物基電解質, 再到具備剛性骨架的無機固體電解質, 電解質的特性決定了其輸運機制. 在液態電解質中, 離子溶劑化之后基于連續流體中梯度濃度進行的傳導表現為攜帶輸運機制. 在聚合物基電解質中, 離子基于可動柔性鏈段的配位進行傳導, 表現為配位間傳遞輸運機制. 在無機固態電解質中, 離子基于剛性骨架所搭建的離子輸運通道進行傳導, 可分為晶體內離子遷移和相間界面處離子輸運. 根據離子遷移與擴散的表現形式, 晶體內離子傳導可進一步分為單離子間隙輸運機制與多離子協同輸運機制.
2.2.1 液態電解質溶劑攜帶離子輸運
鋰離子在液體電解質中的輸運依靠溶劑化Li+在體相電解質中的擴散, 輸運環境隨其遷移過程不斷變化, 即輸運圖像表現為長程無序特性. 電解質中的鋰離子并非獨立遷移, 而是被極性有機溶劑溶劑化, 即借助鋰鹽溶解產生離子與溶劑之間的結合能來克服鋰鹽中的晶格能, 從而實現遷移. 在濃度較低的電解液中離子被溶劑分子包圍, 被完全隔離的鋰離子和陰離子實現快速輸運[3,24,25]. 例如,研究者研究納米LiFePO4與水溶液電解質接觸后形成的“Janus”固液界面, 此界面同時具有類似于體相與溶劑化鋰離子的結構, 有利于鋰離子在界面附近的輸運[26]. 攜帶式離子輸運伴隨著溶劑化與去溶劑化動力學過程, 它們共同描述了離子輸運微觀圖像(如圖3 所示).
在液態電解質中, 離子載流子數(n)與鹽的溶解和解離成比例, 離子遷移率(m)與電解液的粘度(h)相關. 這兩個關聯參數是影響離子傳輸性能的主要因數,并共同描繪了液態電解質離子輸運微觀物理圖像. 這種關系也適用于離子運動通過溶劑化與周圍環境(即溶劑分子)高度耦合的體系. 該體系的極端情況即為固態聚合物電解質, 可視為非水電解質的大分子狀態. 在這種強耦合電解質體系中, 若沒有溶劑化離子的聚合物鏈段的協同運動,離子輸運將無法進行. 離子與溶劑間的偶聯將隨著鹽濃度的增加而不斷增強. 離子輸運圖像更依賴于溶劑分子的本質屬性[10,27].
2.2.2 聚合物中鏈段配位間離子傳遞輸運
傳導離子在聚合物中的直觀輸運圖像由鏈段的移動與配位的傳遞共同刻畫[28-30]. 離子在具有配位的可動聚合物鏈段的“簇擁”下傳遞運動, 即表現為配位間同步傳遞輸運機制(如圖4 所示).
聚合物中的離子輸運包括溶劑化鹽的離解、陽離子與極性基團的配位、離子的鏈段間傳遞等過程. 聚合物中的快速陽離子輸運通常需要聚合物鏈段具備高可動性, 其在非晶態狀態下更有利實現.在低溫下, 聚合物處于結晶或半結晶態; 當溫度升高至Tg時, 結晶聚合物的一部分變為無定形態;繼續提升至熔化溫度(Tm)時, 大部分變得無序并且聚合物熔化. Li+的傳輸主要依賴于聚合物無定形區鏈段的可動性, 因此電池工作溫度應高于Tg.Li+在聚合物電解質中的具體傳輸機制為: Li+與聚合物鏈段上的極性基團配位, 并在電場驅動下隨著鏈段的運動從一個配位點傳輸到下一個配位點. 同時, Li+也有可能通過聚合物電解質中的特殊晶體結構進行傳導, 譬如PEO-LiAsF6中成對的PEO鏈折疊在一起形成圓柱形隧道, Li+與醚氧鍵配合后沿著圓柱形隧道進行傳輸[28]. 因此, 研究普遍認為離子輸運只發生在聚合物電解質的非晶態相, 而結晶階段則存在于具有獨特離子電導率-溫度關系的不同區域.
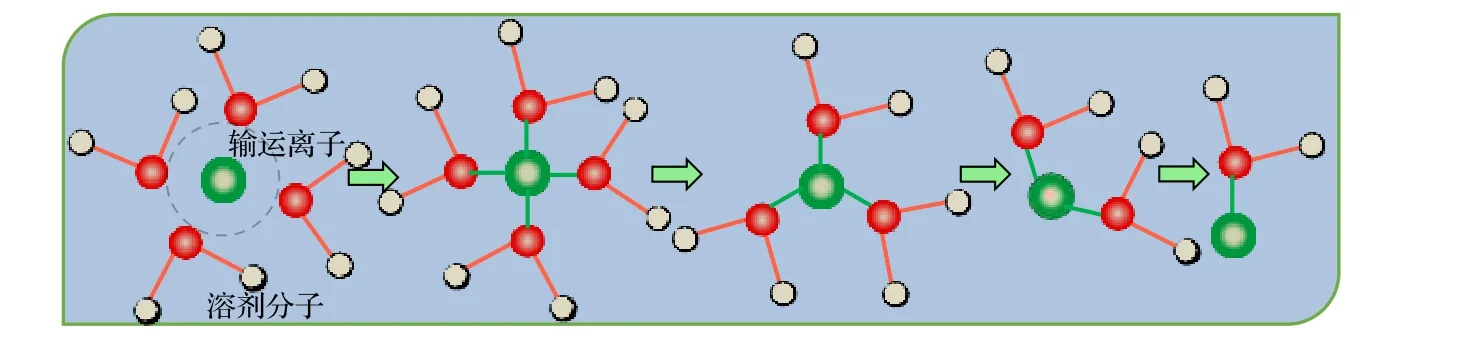
圖3 液態電解質溶劑化與去溶劑化動力學過程中攜帶式離子輸運方式示意圖Fig. 3. The schematic diagram of portable ion transport in the kinetic process of solvation and desolvation of liquid electrolyte.
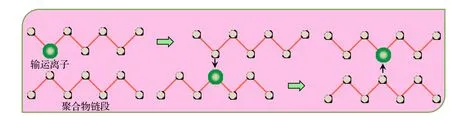
圖4 有機聚合物基固態電解質中離子在配位之間傳遞輸運方式示意圖Fig. 4. The schematic diagram of ion transport between coordination in the organic polymer-based solid electrolyte.
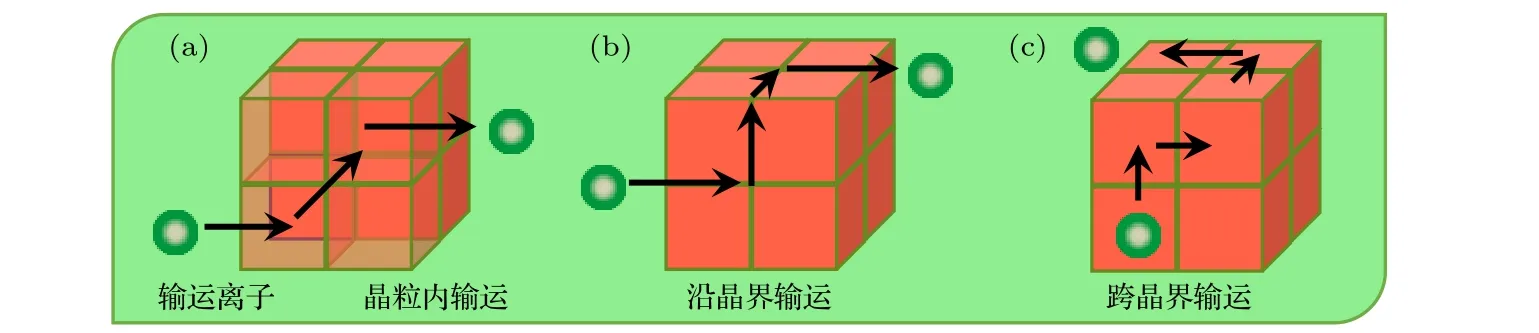
圖5 無機固態電解質中離子輸運方式: (a)傳導離子在晶體內輸運方式示意圖; (b)傳導離子沿晶界輸運方式示意圖; (c)傳導離子跨晶界輸運方式示意圖Fig. 5. The ion transport in the inorganic solid electrolytes: (a) The ion transport in the bulk; (b) the ion transport along the grain boundaries; (c) the ion transport across the grain boundaries.
對于復合固態電解質, 其離子傳導機制主要包括離子在基體中的傳輸、在分散相中的傳輸以及在介于基體與分散相之間的中間相中的傳輸. 由于中間相具有較嚴重的結構不匹配性及較強的化學勢梯度, 因此其結構會發生較大的變化, 界相結構與基體和分散相均具有顯著差異, 導致離子輸運微觀物理圖像不統一, 從而深刻影響離子電導率. 一般來說, 基體的離子傳導對復合固態電解質整體離子電導率至關重要, 而分散相的加入可能改變基體的相穩定性及其微觀結構, 兩者共同決定了整體離子導電性. 這種現象通常發生在聚合物基復合固態電解質, 在晶體基復合固態電解質也會出現. 前者的離子輸運高度依賴于聚合物的微觀結構[7,31].
2.2.3 無機固態電解質中離子通道輸運
無機固態電解質中離子輸運特征與液體電解質和聚合物電解質均有所不同. 由于無機固態電解質骨架具有一定的剛性, 其輸運圖像具有一定的機械特性, 從而離子輸運圖像可由電解質的本質屬性直接勾勒. 盡管目前暫未完全了解無機固態電解質中的離子遷移機理, 從物質結構角度大致可分為兩類: 晶體中離子輸運和相間界面離子輸運(如圖5所示)[8,32,33]. 根據不同界面擴散的物理模型, 界面離子輸運通常分為: 離子沿晶界輸運與離子跨晶界輸運(如圖5(b)和圖5(c)所示)[34]. 界面效應的離子輸運特性可改善鋰負極與固體電解質界面阻抗,以及可提高鋰負極表面固體電解質界面(SEI)中離子輸運效能. Pan 等人[35]通過LiF/Li2CO3界面缺陷形成的空間電荷效應耗盡電子載流子而提高離子載流子積累的方式搭建可供離子輸運的人工SEI. Ren 等人[36]通過第一性原理計算已證實LiF-SEI 的界面區域的變化對離子擴散能壘影響較小. Li 等人[37]為了減小固體電解質與鋰金屬負極之間的界面阻抗, 在兩者之間引入LiF 作為過渡界面, 有效利用界面相快速輸運鋰離子性能來降低固-固界面阻抗.
1) 無機固態電解質中間隙輸運
離子輸運發生在具有固定形式的晶格中, 離子在晶格中占位決定其勢能, 而離子在遷移過程中發生的勢能變化決定其活化能與遷移路徑. 傳導離子可以在體相固態電解質中沿相鄰位點跳躍, 即輸運離子克服瓶頸限制使骨架畸變從而實現間隙擴散(如圖6(a)所示); 或在選擇輸運通道的過程中優先找尋晶體中相鄰的空位或缺陷位置進行空位輸運(如圖6(b), 其中方形表示空位). 由于離子通過躍遷的方式在骨架離子的相互作用驅動下在相鄰位點間遷移[38-40], 因此離子輸運圖像的描述因子更側重于傳導介質的結構形式, 而輸運過程的能壘可為刻畫物理圖像提供定量信息.
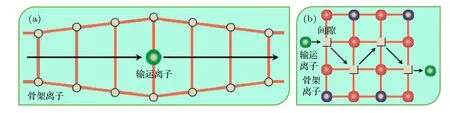
圖6 無機固態電解質中晶體內離子間隙擴散輸運方式:(a)離子直接在間隙中遷移示意圖; (b)離子在空位之間遷移示意圖Fig. 6. The ion interstitial diffusion transport in the inorganic solid electrolytes: (a) The interstitial ion transport; (b) the vacant ion transport.
2) 無機固態電解質中離子換位協同輸運
對于無機固態電解質中離子協同輸運機制的研究, 2012 年, 我們[21]首次闡明間隙Li+在主要成分為Li2CO3的固體電解質界面膜(SEI)中的Knock-off(間隙Li+和晶格Li+協同交換位置)協同輸運機理. 2017 年, Mo 等人指出處在高低能位離子之間的強庫侖相互作用[類似于勢能的相互轉化]是離子協同輸運的關鍵, 可降低離子遷移勢壘[41]. 在多離子協同躍遷中, 各個離子所處的位置能量不盡相同. 當相鄰離子一起運動時, 高能量位置的離子向下運動, 部分抵消低能量位置離子向上運動的能壘. 因此在多個離子的共同運動下, 協同躍遷表現出更低的能壘(如圖7 所示). 在不同的無機固態電解質(Thio-LISICON Li10SiP2S12,Li1+xAlxTi2—x(PO4)3, Li14P2Ge2S16—6xOx, NASICON等)中, 通過離子協同傳輸機制描述其輸運特性,目的在于提高離子電導率[23,42-45]. 最近,我們開發了一套“配位多面體”方法分析離子協同跳躍的程序對協同程度定量化進行刻畫,藉此首次揭示單斜Na3Zr2Si2PO12結構中的Na5 位置,高能態的Na5 位點作為交叉位點使離子得以快速輸運,Na+離子傳導主要通過多離子的協同跳躍機制發生,Na+離子濃度的增加導致庫侖排斥力增加,并激活更多的協同輸運[45]。實現離子協同輸運程度的定量化刻畫對于指導新型高導電性固態電解質材料的設計與改性有著重要意義.
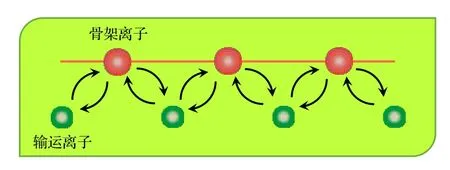
圖7 無機固態電解質中輸運離子與骨架離子換位協同輸運示意圖Fig. 7. The concerted and coordinated diffusion of transport ion and skeleton ion in the inorganic solid electrolytes.
協同輸運機制適用于骨架幾何形狀突出的離子導體, 其本質為間隙離子與骨架離子之間以及輸運離子之間的相互作用改變了離子遷移方式, 降低了輸運能壘. 早期被提出的“Knock-off”機制[21], 以及“concerted migration”機制[41], “cooperative”機制[42], “collective”機制[43], “Interstitialcy”機制[44],“correlated migration”機制[23,45]等均為針對協同輸運所提出的相關描述. 然而, 對于骨架幾何形狀并不突出的離子導體, 其輸運行為更適合“Grotthuss”機制[46]. 在離子協同輸運過程中存在多種協同運動方式, 例如: 同種離子間協同(如Li+-Li+或Na+-Na+[23,45])、異種同價離子間協同(如Li+-Na+[47])、我們最近提出的異種異價離子間協同(如Na+-Zn2+[48])及異種電荷離子協同(如Co 離子與O 離子[49]). 協同輸運過程中又存在同種離子與不同離子分別沿同一方向與不同方向的協同運動方式均影響離子輸運的物理圖像[48]. 在同種離子協同輸運過程中, “協同離子”與“輸運離子”分別占據高低能位后通過庫侖作用力互相推動協同運動, 運動勢能進行互補降低系統總激活能[23,45]. “協同離子”與“輸運離子”的占位與濃度決定了局域平衡構型的形成, 增加低能壘協同運動,使得協同跳躍率升高, 進而提高了擴散系數. 由此,協同跳躍率可定量描述協同輸運程度[23,45]. 正是基于儲能材料中也會存在異種異價遷移離子協同輸運的方式,我們率先發現NASICON 結構NaV2(PO4)3作為鋅離子電池電極在Zn2+脫嵌過程中可通過Na+/Zn2+離子協同輸運“激活”M1 位置的方式,實現良好的電池充放電循環性能[48]. 需要指出的是, 盡管離子的換位協同輸運機理在計算上被提出廣泛存在于各類無機固體電解質中, 但要實現實驗上的直接觀測還是一個挑戰.
3 離子輸運微觀物理圖像表達
3.1 液態電解質離子輸運
液態電解液具有較高的離子電導率、較寬的電化學窗口、較高的穩定性, 與正負極的物理接觸良好, 因此廣泛應用于各類電池體系. 液態電解質通常由溶劑分子、陰離子以及溶劑化的鋰離子組成,并且遵循典型的“溶劑化離子”電解液模型, 因此,可將其定義為“Li+溶劑化的電解液”. 整體來看, 離子輸運與去溶劑化動力學過程同步進行. 由于鋰離子嵌入電極與其去溶劑化同步進行, 如果參與溶劑化的溶劑分子之間存在作用力, 那么去溶劑化分子的電荷并非均勻分布, 因此具有較高反應活性. 這些高能態溶劑分子在電極表面的穩定性差, 容易發生界面副反應導致電化學穩定窗口變窄[50]. 因此,離子輸運與去溶劑化共存的過程將對電池的容量和循環壽命產生不利影響.
另一方面, 電解質研究的主要目的之一提高電導率和鋰離子遷移數[51,52]. Li 鹽(如: LiPF6)解離為陽離子(Li+)和陰離子的能力與溶劑的介電常數密切相關, 通常介電常數越高溶劑化能力越強[53]. 鋰離子被溶劑分子完全包圍發生溶劑化可以減少陰離子的影響. 而較大陰離子可以促進負電荷較好分布,有利于防止離子配對,因此對電導率和溶解度較為有利[54]. 此外, 低粘度也有助于離子輸運速度的提高[55]. 為滿足電池系統的正常工作,電解質的室溫電導率通常至少需達到10—3S·cm—1[56].綜上所述, 因涉及較多影響因素(如黏度, 鹽濃度,溶劑化, 離子締合和離子與溶劑的相互作用), 液態電解質中的離子遷移機理仍難以直觀理解.
3.1.1 液化氣電解液離子輸運
液態電解質的研究過程中, 一系列有效手段被陸續提出. 譬如, 研究者針對液化氣電解液, 通過添加共溶劑提高了液態電解質中鋰鹽溶解性, 快速溶解的鋰鹽提高了鋰離子的遷移率, 明顯改善離子電導率[57]. 發現液化氣電解液的團聚程度隨溫度變化明顯, Li+自由離子濃度隨溫度降低而增加, 且具有較快的擴散速度, 對電解質中離子輸運起到促進作用, 而TFSI—自由離子濃度在各溫度都趨近于無. 因此液化氣電解液具有極高的低溫導電性(2.8 mS·cm—1, —60 ℃)和高Li+遷移數(tLi+≈0.79).
3.1.2 溶劑化電解質離子輸運
高濃度“鹽包水”電解質可以提供較寬的電化學穩定性窗口, 但是由于缺乏滿足水溶性條件的鋰鹽, 通常基質只能選擇有毒且成本高的有機酰亞胺. 為此, Lukatskaya 等[58]提出混合陽離子法, 即利用高溶解度的乙酸鉀, 實現鋰和乙酸鉀的共晶混合物中水與陽離子摩爾比為1.3 的“鹽包水”狀態.由共享的水分子組成陽離子溶劑化結構中, 陰離子以配體形式存在. 該鹽包水混合陽離子溶劑化結構中水-水氫鍵的破壞和強離子相互作用協同完成離子輸運行為, 且乙酸鉀基高濃度電解質可以提供與酰亞胺基電解質相同的擴展電壓范圍優勢.
另一類有希望的液態電解質材料是離子液體.作為室溫熔融鹽[59], 它們具有一系列獨特優勢, 如不燃性、低蒸氣壓、高熱穩定性、高電化學穩定性、低毒和高離子含量[60-62]. 然而, 離子液體的粘度比常用的有機液體電解質高一到兩個數量級, 以至于其離子電導率低三到四個數量級[61]. Zhang 等人[63]在含LiFSI 的醚類(DME)電解液中加入一定量的LiNO3, 通過可自分解的LiNO3對FSI—陰離子調控, 使其完全分解, 從而使得鋰離子在含有大量LiSOx, LiF 和LiNxOy的SEI 中均勻快速傳輸. 由此, 研究者基于對LiNO3調控溶劑化層中陰離子策略設計出鋰金屬電池中含LiClO4的電解液[63].
針對電解液基于去溶劑化的輸運過程中出現的溶劑分解、電化學穩定窗口窄、電解質與電極界面不穩定等現象, 在保證離子電導率的基礎上,Chang 等[64]將鋰離子的去溶劑化過程從高反應活性的電極表面提前轉移到穩定且絕緣的金屬有機框架(MOF)孔道內(2.9 ?), 獲得了一種特殊的“Li+去溶劑化(醚基)電解液”. 該液態電解液僅由非活性的“冷凍狀”溶劑分子(“frozen-like” DME)和由去溶劑化后的鋰離子與陰離子構成的類晶體狀鹽溶質(“crystal-like” salt)組成. 基于此電解質的電池在充放電過程中, 僅有裸鋰離子嵌入電極材料表面, 因而抑制高能態的溶劑分子與高反應活性的電極表面直接接觸, 避免常規液態電解液的固有缺陷.
在稀溶液中, LiTFSI 離子對主要通過介質機理移動. 超濃縮溶液可能會破壞介質的主導地位,導致重復的離子締合-解離過程, 從而轉化為結構擴散. 但是, 對于陰離子相對較大的情況, 環丁砜(SL)溶劑在—SO2基團中具有兩個間隔很近的溶劑化氧(如圖8 所示), 從而使Li+能夠以與移動SL分子大小相同的時間尺度交換. Li+和溶劑在鹽溶液中(SL/Li = 8.33)進行擴散, 在超濃縮鹽溶液中(SL/Li = 2.56), Li+擴散比SL 擴散更為突出[10].在超高鹽濃度溶液中的離子輸運機制接近固態電解質中離子傳導方式, 輸運離子與溶劑分子重復成鍵與斷鍵過程跳躍遷移[65,66].
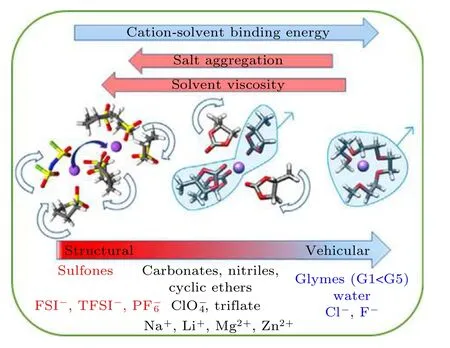
圖8 電解質中主導離子輸運微觀物理圖像的因素由結構主導作用到介質主導作用的演變過程[10]Fig. 8. The evolution process between structural and vehicular effect the microscopic physical image of the contribution to the ion transport in the electrolytes[10].
3.1.3 固態冰電解質離子輸運
通過研究固態冰中離子傳導行為, 研究者發現了低溫固態電解質離子輸運特性[67]. 采用快速冷卻法將硫酸鹽、硝酸鹽、氯化物的水溶液轉變為多種固態冰材料后, 能夠傳導不同的金屬離子(Li+,Na+, Mg2+, Al3+, K+, Mn2+等). 其中, 溶解Li2SO4的冰系固態電解質在—9.2 ℃下的鋰離子電導率達到6.4 × 10—4S·cm—1. 以溶解1 mol/L CuSO4的冰為模型進行第一性原理計算, 發現存在2 條可供Cu2+在冰晶體結構中輸運的路徑, 其擴散勢壘分別為0.35 和0.44 eV. 通過交流阻抗譜(EIS)、直流電沉積、冷凍透射電鏡(cryo-TEM)等實驗手段,證明這類固態冰材料的晶格中存在金屬離子缺陷,因此能夠在外部電場的驅動下進行離子遷移. 利用快速冷卻相變過程保存溶液中的離子并具備傳輸能力, 這一簡單易行的方法可擴展至有機溶液、水凝膠等體系, 也可通過使用不同的金屬鹽擴充傳輸離子種類. 此外, 冰系固態電解質能夠在較低溫度下工作, 這為低溫電化學器件的設計提供了新的發展思路. 譬如在約—8 ℃時, 研究發現各種硫酸鹽的離子電導率高達10—3S·cm—1, 其中Li+和其他堿性離子的遷移通過冰晶格的離子跳躍機制實現.
3.2 聚合物基電解質離子輸運
聚合物電解質具有一定機械強度, 且可較好匹配正負極以形成較低阻抗界面, 同時大大提高了電池體系的安全性, 因而被廣泛關注. Fenton 等[68]最早發現聚環氧乙烷(PEO)與堿金屬鈉鹽絡合物具有離子導電性; Armand 等[69]率先將聚合物電解質應用于電池體系, 由此開創了新型聚合物電解質的設計與離子輸運機制的研究方向. 聚合物電解質(膠凝聚合物和無溶劑聚合物)的離子導電機理與無機晶體材料和液體型電解質的離子輸運機制大不相同. 在無溶劑的聚合物電解質中僅在Tg之上顯示出快速離子傳導. 因此, 低Tg聚合物如PEO(聚環氧乙烷,Tg約為—50 ℃——57 ℃)已成為無溶劑電解質的重要聚合物主體, 無定形化研究可以增加其離子電導率. 由于低分子量溶劑在聚合物中的擴散以及聚合物鏈段的運動, 膠凝聚合物顯示出比無溶劑聚合物更快的離子傳導能力[29].
3.2.1 骨架結構聚合物基電解質
為降低固態電解質與電極間界面阻抗的影響,通常聚合物基電解質多以具有柔性結構且與電極較好界面接觸的聚丙烯腈(PAN)和聚甲基丙烯酸甲酯(PMMA)等為基體. 以PAN 或PMMA 等為基體吸附有機增塑劑形成的凝膠聚合物電解質(GPE)中的離子輸運主要依賴液態增塑劑[70]. 而在以PAN 為支撐骨架的塑晶基固態電解質中形成輸運網絡為離子擴散提供了快速通道[71]. 通過枝接在聚合物骨架上的陰離子形成單離子聚合物電解質減少陰離子的遷移來提高Li+遷移數, 是提高聚合物基電解質離子電導率的較優選擇. 但是, Li+反而受到被固定的陰離子基團較強的牽制作用[72].為獲得較理想的離子電導率, 研究者通過調整搭建特殊結構的聚合物基電解質來調控其中的離子輸運模式, 如交聯網絡結構與復合結構聚合物基電解質.
3.2.2 交聯結構聚合物基電解質
Yu 等[73]搭建了三類離子傳導方式的物理模型: 動態單離子傳導、非動態單離子傳導、非動態非單離子傳導. 并設計了三種化學結構類似的聚合物電解質, 通過離子輸運特性系統地研究了SEI 性質對于金屬鋰電池特性的影響: 基于AlO4四面體陰離子和Li+的交聯網絡Al-FTEG; 基于BO4四面體陰離子和Li+的交聯網絡B-FTEG; 基于SiO4四面體的共價交聯網絡Si-FTEG. 在離子輸運過程中, 陰離子不僅作為交聯中心, 而且成為鋰離子的對離子, 還能通過化學鍵的相對強弱來調控宏觀的動態流動性. Li+離子離開某一個陰離子中心之后, 會被氟代配位鏈上的F 基團暫時穩定, 形成輸運中間體從而躍遷到下一個陰離子中心.
具有較高離子電導率的醚類電解質高電壓環境易受氧化導致不穩定, 不利于匹配高能量密度高壓正極材料. 為實現單一電解質中高離子電導率與高氧化穩定性的統一, 研究者提出一種新型氟化醚聚合物電解質, 即通過氟化核心與醚“端基”共價鍵合的共同作用實現離子輸運[74]. 同時, 通過模塊化的方式改變醚基的長度和類型以及氟化鏈段的長度, 發現具有較長的醚基團和較短的氟化鏈段時,離子電導率高達2.7 × 10—4S·cm—1(30 ℃), 同時氧化電壓高達5.6 V. 隨著醚段的增加和氟段的縮短, 氟化醚中的離子電導率會進一步增加.
基于常規聚環氧乙烷(PEO)的固態聚合物電解質往往離子電導率和遷移率較低, 因此研究者引入交聯的聚四氫呋喃(xPTHF)以構筑松散配位的高性能聚合物電解質(xPTHF10)[75]. 與常用的xPEO體系相比,xPTHF 體系具有0.53 的高轉移數, 良好的電化學穩定性和更高的離子電導率. 此外,xPTHF10在234 ℃下表現出良好的熱穩定性, 并在全固態磷酸鐵鋰電池中展現出優異性能, 在70 ℃和1C 倍率下的電池比容量達到162 mAh·g—1. 通過分子添加劑(例如DMF 和PC)可進一步調節Li+配位環境以改善離子輸運, 譬如xPTHF5DMF 2∶1基固態聚合物電解質的室溫離子電導率可以達到1.2 × 10—4S·cm—1.
固態電解質的另一方面挑戰在于機械性能和離子電導率的協調匹配. 研究者針對性地設計了一種具有雙共價和動態氫鍵交聯的彈性鋰離子導體,在擁有高機械彈性的同時保證了室溫離子電導率.其中聚環氧丙烷彈性體(ePPO)通過靜電共價鍵的組合提供彈性, 酰胺基團之間的動態氫鍵可消除應力[76]. 聚醚胺前驅體轉化為氫鍵合酰胺后, PPO主鏈形成無定形域而不是由PEO 形成結晶域. 同時通過線性二胺增加交聯的分子量, 降低材料的機械模量, 并使純聚合物的應變能明顯提升. 應用該電解質的Li/LiFePO4電池在室溫下能夠以152 mAh·g—1的高陰極容量運行300 次, 且對機械沖擊展現出良好的耐受性. 這種雙交聯設計不僅為固態電解質提供了強大的機械彈性, 同時達到當前聚合物基電解質離子電導率的最好水平. 沿著協調聚合物的離子電導率與機械強度的思路, 研究者進一步發展出一種超分子鋰離子導體[77]. 低Tg的聚醚主鏈單元保證了室溫下達到(1.2 ± 0.21) × 10—4S·cm—1的高離子電導率; 同時高機械強度來源于動態鍵耦合的2-脲基—4-嘧啶酮(UPy)主鏈單元, 韌性達(29.3 ± 1.4) MJ·m—3. 具有一定柔性的聚合物電解質易通過調控配位環境優化離子傳遞的模式.在聚合物基固態電解質中構建交聯網絡可以為離子輸運提供快速擴散通道, 有效提高電解質的離子電導率.
3.2.3 復合結構聚合物基電解質
為進一步提高聚合物電解質的機械強度, 復合固態聚合物電解質被發展出來. 復合固態電解質具有堅固、不易燃的主體, 并采用具有垂直排列納米通道的固態聚合物電解質填料. 高模量主體可以抑制枝晶滲透, 而對齊的通道增強固態聚合物電解質填料的離子導電性可以較好地描述離子輸運微觀物理圖像. 復合電解質的超薄特征和主體-填料性能使得全電池具有極大的柔韌性、低電解質電阻和潛在的高能量密度. 基于此思路, 研究者采用高模量的納米多孔聚酰亞胺(PI)主體和PEO/鋰雙(三氟甲磺酰基)酰亞胺(LiTFSI)聚合物電解質進行復合設計, 所獲得的PI/PEO/LiTFSI固體電解質中的超薄多孔PI 基質厚度為8.6 μm[78]. 相應的全固態電池的能量密度(246 Wh·kg—1)與液體電解質電池相當. 而垂直通道的設計可顯著提高離子電導率, 30 ℃時為2.3 × 10—4S·cm—1. 同時具有液態流動性與晶體有序性的納米結構復合聚合物基離子導體常被稱為液晶電解質, 其較好的離子輸運性能被廣泛關注. 由于離子型與非離子型液晶分子復合在電解質中形成特殊的介晶納米結構, 在所形成柱狀相、近晶相及雙連續立方相的結構中存在1D,2D, 3D 離子快速輸運通道[79-81].
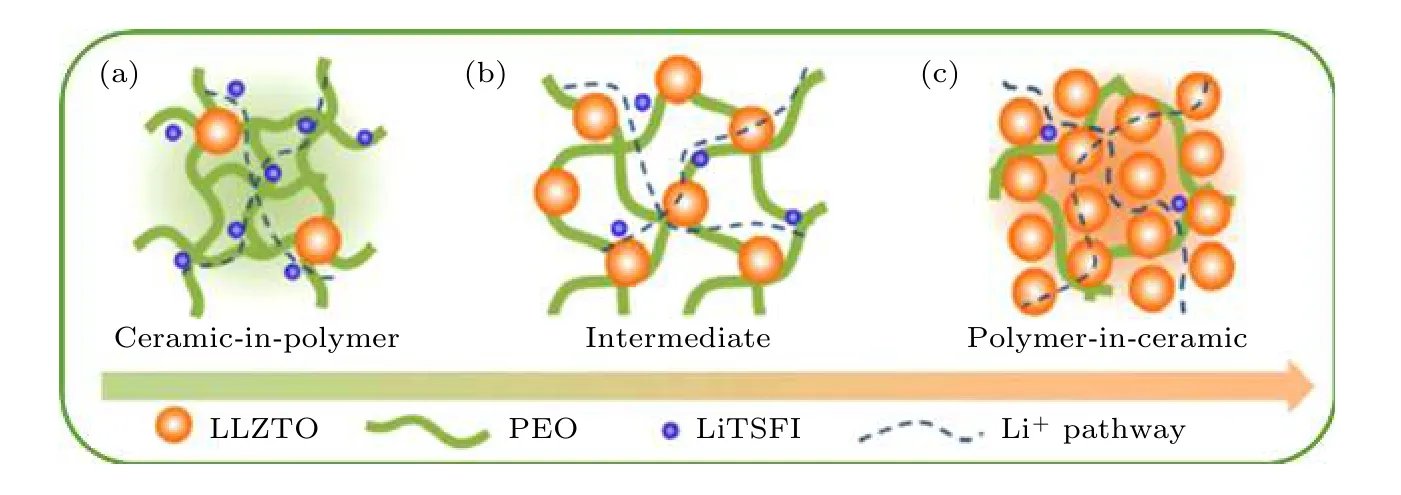
圖9 “聚合物陶瓷”和“陶瓷聚合物”電解質系統中可能的離子傳輸機制[82]Fig. 9. The schematic diagram of possible ion transport mechanisms in "ceramic-in-polymer" and "polymer-in-ceramic" electrolyte system[82].
復合固態電解質的研究過程存在從“聚合物中的陶瓷”到“陶瓷中的聚合物”模型的演變, 即物理形態從柔性載體向剛性骨架固態電解質的轉變(如圖9 所示). 具體地說, PEO 聚合物基體與負極的鍵合可以改善電解質和電極的接觸, 有利于固/固界面相容性. LLZTO提供的Li+傳輸通道有助于增強Li+傳導能力[82,83]. 具有PEO 鏈段的剛性LLZTO 顆粒可提供機械堅固且穩定的框架, 以抑制鋰枝晶生長. 而且LLZTO與鋰金屬的穩定化學和電化學性能使電解質/鋰界面達到穩定狀態.研究者將含有PVDF 和LLZTO的分級多孔復合固態電解質涂覆在商品化聚丙烯隔膜上形成復合隔膜電解質. 其中, PVDF 與LLZTO之間的相互作用在界面上形成的連續鋰離子通道誘導來自聚丙烯隔膜的不均勻鋰離子流重新分布[84], 從而促進離子輸運.
3.3 無機固態電解質離子輸運
多數由離子鍵和共價鍵形成的固體中并不存在明顯的離子輸運. 在高溫條件下晶體缺陷濃度增大, 離子才表現出遷移行為, 具有相對可觀的離子電導率. 然而, 被稱為固體電解質或快離子導體的固體中離子較容易遷移, 通常要求較高的室溫離子電導率(10—3—101S·cm—1), 較低的活化能(0—0.5 eV)[85,86]. 盡管在早期的研究中將離子電導率接近(或在某種情況下超過)熔鹽和電解質溶液的一類固體材料定義為固體電解質[85,86]; 但隨著其在能源、冶金和化工等領域有著越來越廣泛的應用, 僅能傳導離子而對電子絕緣的(衍生)固態物質均被稱為固體電解質. 因為這類具有特殊晶體結構的固體中存在可供離子輸運的開放隧道或開放層, 結構中的陽離子或陰離子并非限制在特定晶格位置, 而是可在結構中自由遷移. 因此, 固體電解質在結構和性質上介于具有規整三維結構且離子不可移動的正常晶態固體與無規則結構且離子可移動的液體電解質之間.
固態電解質與液態電解質相比具有許多優勢,包括設計簡單、自然密封、耐沖擊和振動、耐壓力和溫度變化、更寬的電化學穩定性和更好的安全性. 然而, 這類材料仍然受限于相對較低的離子電導率. 無機固態電解質中的空間有序晶態陶瓷相與長程無序非晶玻璃態離子輸運微觀物理圖像主導因素更取決于介質的通道特性及輸運環境各離子間作用力. 如前所述, 無機結晶化合物中的離子傳導是由于流動離子在周圍電勢中的能量有利位置之間跳躍導致的, 而周圍離子的運動僅為移動離子提供了通過晶體框架中的通道移動的活化能, 因此離子電導率一般較低.
3.3.1 氧化物型固態電解質
1) 石榴石型固態電解質
石榴石型Li7La3Zr2O12(LLZO)固態電解質因具有高離子電導率(10—4—10—3S·cm—1)、對鋰金屬的良好的穩定性、較寬的電化學窗口以及相對環境友好性, 自2007 年發現以來便被認為是固態電池最有前途的電解質之一[87]. Wang 等[11]和Du 等[88]全面論述了石榴石型固態電解質材料和相應固態電池的發展歷程, 其中石榴石型氧化物鋰鑭鋯氧(LZZO)格外受到關注. 整體來看, 石榴石型氧化物離子電導率介于硫化物和聚合物之間. 研究發現, 室溫離子電導率達到10—3S·cm—1的陶瓷片的厚度需要小于500 μm, 室溫離子電導率達到10—4S·cm—1的柔性膜厚度則需小于50 μm. 研究者針對Li7La3Zr2O12中的離子遷移過程采用傳統粉末衍射結合最大熵法(MEM)進行描述, 揭示了結構中存在一個連續的三維移動通道, 即鋰離子通過24d-96g-48g-96h-24d 路徑在結構中遷移[9,89]對LLZO 在正負極界面處的輸運進詳細分析, 發現Li 與LLZO 界面輸運需要高離子相融性中間層復合正極與LLZO 陶瓷片之間則需要高離子電子共導通的相融性中間層相關的離子輸運微觀圖像描述尤為重要[90].
2) NASICON 型固態電解質
Na1+xZr2SixP3—xO12(0 ≤x≤ 3)由PO4(SiO4)四面體和ZrO6八面體通過共角相連組成結構框架, 可兼具固體電解質和電極功能, 因此該框架結構的化合物被統稱為NASICON (antrium superionic conductors)[91,92]. 其中, Na3Zr2Si2PO12在1976年被Hong[91]和Goodenough 等[93]首次提出, 其作為Na+固態電解質在300 ℃時的離子電導率達到0.2 S·cm—1. 摻雜結構Na3Zr2Si3PO12的室溫Na+離子電導率也已提高到3.4 × 10—3S·cm—1[94].由于通過用Li+取代Na+并在Zr 位進行摻雜等改性方法, 可以得到具有較高離子電導率的鋰離子固態電解質(如 Li1.3Al0.3Ti1.7(PO4)3, 室溫Li+離子電導率可達7 × 10—4S·cm—1[95]).
NASICON 結構化合物中具有較多的傳導離子可占據位置, 傳導離子在這些晶格位、間隙位或晶格-間隙位之間涉及到多個離子的協同輸運, 因此可能具有更低的輸運勢壘. Lin 等[96]認為NaZr2(PO4)3體系中用Si 取代P, 會增加Na+濃度, 而后增加的Na+會填充在Na2 位. 由于Na1位全占據, 位于Na2 位的Na+將會與位于Na1 位的Na+采取協同方式輸運, 即位于Na2 位的Na+躍遷至Na1 位, 同時由于靜電排斥作用, 位于Na1位的Na+將跳至下一Na2 空位, 依次循環實現離子傳輸. He 等[41]也發現Li1.3Al0.3Ti1.7(PO4)3體系中, Li+通過協同輸運的方式可以降低勢壘. 相似地,MTi2(PO4)3(M= Li, Na)中由溫度或傳導離子濃度等因素影響產生的間隙位M離子將以協同方式進行輸運, 以實現MTi2(PO4)3(M= Li, Na)的快離子傳輸[97].
3)β-Al2O3固態電解質
β-Al2O3一直被用作高溫固體電解質, 但由于離子電導率極高的優勢也可在接近室溫條件下工作[98,99]. 以Na+離子或其他堿金屬離子為載流子的β-Al2O3可在較低的溫度下表現出較高的離子電導率(10—1S·cm—1)[100], 接近液態電解質水平. 在β-Al2O3中, Na+離子在Na-O 層狀亞晶格的二維通道中遷移. 為了改善β-Al2O3中離子輸運圖像,研究者希望制備具有三維通道結構的化合物, 從而使Na+離子輸運圖像表現為各向同性. 從β-Al2O3的晶體結構來看, Na+離子在其中的擴散在垂直于c軸的二維空間發生, 并未在平行于c軸的方向通過密堆積的氧層運動, Li+離子的擴散方式與Na+離子相同. 室溫下Na-β-Al2O3可實現與Na 金屬的低界面電阻, 使短路臨界電流密度提高到12 mA·cm—2[101]. 在Na-β-Al2O3中, 鈉氧層具有一定厚度(約4.76?), 因此受輸運陽離子的尺寸效應影響明顯: 若輸運陽離子太小(如Li+離子), 其將位于Na-O 層的一側, 從而受靜電引力影響導致遷移能較大; 若輸運陽離子較大(Na+離子), 上下兩層電子云斥力使其保持在中間位置, 遷移時無需附加能量, 即具有較低的活化能和較高的電導率; 若輸運離子更大(如K+離子), 其受到上下氧層的斥力更大, 遷移所需較高活化能, 所以擴散系數與電導率反而較低.
3.3.2 硫化物型固態電解質
硫化物固體電解質Li10GeP2S12(LGPS)的離子電導率接近甚至高于液態電解質的離子電導率, 其中文獻報道的LGPS 離子電導率可達2.2 mS·cm—1[102]和12.0 mS·cm—1[103], LiSiPSCl 的離子電導率甚至可達到25.0 mS·cm—1[104]. 因此, 硫化物固態電解質在實現全固態電池方面被寄予厚望.
Li10GeP2S12(LGPS)是第一個被報道室溫鋰離子電導率超過液態電解質水平的固態電解質材料(12 mS·cm—1)[103]. LGPS 空間群為P42/nmc, 屬于四方晶系. 由四面體構型的(Ge0.5P0.5)S4和八面體構型的LiS6組成結構框架, (Ge0.5P0.5)S4與LiS6共邊, 沿c軸形成一維鏈, 一維鏈與鏈之間通過四面體構型的PS4與八面體構型的LiS6共頂角互相連接. 相應的一維離子輸運通道由四面體構型LiS4中的16h 和8f 位點形成, 其中共邊連接成一維四面體鏈, 鏈與鏈之間通過四面體LiS4共頂角連接. 研究發現, 鋰在16h 和8f 位置的熱振動表現出高度的各向異性, 鋰從16h 和8f 位置分別向2 個16h 位置之間和16h 與8f 位置之間的間隙位置遷移. 這表明沿c軸存在一維傳導通路, 且進一步發現鋰離子沿傳導通路的占位分布均勻. 因此,硫化物無機電解質離子輸運的微觀物理圖像表現出了超離子導體的特征[105]. 硫化物無機固體電解質中離子輸運所表現出的各向異性物理圖像, 與其相似的輸運圖像在聚合物固體電解質中也出現[73].但是, 前者主要提高了電解質的離子電導率, 后者則改善了SEI 的生長模式, 進而減少枝晶生長.
研究者基于硫化物固體電解質, 提出了在固體電解質中引入多種共存陰離子, 并設計了氧硫化物固體電解質LiAlSO 材料. 通過基于晶體結構預測方法的高通量計算, 確定了該材料的晶體結構, 并研究了其熱力學穩定性、動力學穩定性和離子輸運性質. 計算結果顯示該化合物在a軸方向具有很低的鋰離子遷移勢壘, 屬于快離子導體. 通過BVpath程序計算解析LiAlSO 中的鋰離子輸運通道, 發現Li+沿a方向由間隙離子與晶格位離子協同運動的遷移過程, Li+沿a軸方向空位遷移過程, 以及Li+沿c方向間隙離子遷移過程共同組成離子輸運微觀物理圖像[106].
通過研究聚陰離子在以α-Li2SO4基和α-Na3PO4基的無機塑晶電解質和Li10MP2S12(M=Si, Ge, Sn)電解質中的旋轉運動效應, 發現較大尺寸聚陰離子的取代雖擴大了骨架晶格, 但抑制了聚陰離子的旋轉運動, 導致離子電導率下降; 而在以聚陰離子的旋轉運動為主導效應的Li10MP2S12(M= Si, Ge, Se)電解質的離子電導率被提高[20].關鍵在于如何有效激活聚陰離子旋轉效應. 含可旋轉聚陰離子的固態材料(例如)由于具有獨特的傳輸行為而構成一類特殊的離子導體, 其中旋轉的聚陰離子可促進相轉變為無序相, 并顯著提高陽離子電導率. 研究者曾通過高氫配位氫化物激活聚陰離子旋轉效應, 將含有MoH39結構的復雜過渡金屬氫化物Li5MoH11的室溫離子電導率提高到7.9 × 10—2S·cm—1[107]. 對于玻璃態電解質75 Li2S—25P2S5, 鋰離子遷移表現為協同輸運與陰離子旋轉效應動態耦合的“paddlewheel”機制. 對于復雜陰離子且原子結構不具有遠程共價網絡的玻璃電解質, 通過促進“paddlewheel”動力學來增強低溫下的陽離子遷移率也是一種較好的策略[108].
3.3.3 鹵化物型固態電解質
金屬鹵化鋰固態電解質Li3TMHa6(TM= Y,Er, Sc, In, ···,Ha= F, Cl, Br)具有高室溫離子電導率(>10—3S·cm—1)、寬電化學穩定窗口以及與氧化物正極良好的兼容性等諸多優勢, 因而在全固態鋰電池領域備受關注[109].
Asano 團隊[110]提出Li3YCl6(LYC) 和Li3YBr6(LYB)金屬鹵化鋰固態電解質中鋰離子輸運路徑,如圖10(a)所示. 晶體中Y 與鹵素元素組成穩定八面體結構, 同時Y3+離子可以提供正離子空位, 因此有利于鋰離子傳導. Wang 等[111]進一步發現LYC 和LYB 中, hcp 和fcc 陰離子晶格中Li+的擴散路徑為低勢壘空位點在八面體位點間躍遷. 傳導離子的擴散在fcc 的LYB 中表現為各向同性, 而在hcp 的LYC 一維c-通道中受缺陷影響表現為各向異性(如圖10(b)). 因此Li+可在低遷移能壘的鹵素元素陰離子亞晶格中快速傳導(10—3S·cm—1),且無需激活主晶體中鋰離子, 展現了良好輸運特性. Schlem 等[112]研究Li3ErCl6和Li3YCl6的結構和輸運行為, 發現Er/Y 被無序地排列到新位置的晶體結構中導致陽離子缺陷的出現, 可以極大地改善離子傳輸性能(如圖10(c)), 即無序度的增加導致活化能的降低和離子電導率的提高. 對于LixScCl3+x, 發現x值增加導致鋰離子濃度增加、晶體中空位濃度下降, 因此Sc 對傳導離子的阻隔效應減小, 從而增強了鋰離子遷移調節能力. 整體上,LixScCl3+x固態電解質的離子電導率取決于晶體中Li+濃度和總空位濃度[113]. 基于此輸運機制的ccp 陰離子堆積結構的Li3ScCl6固態電解質, 其鋰離子遷移路徑如圖10(d)所示.
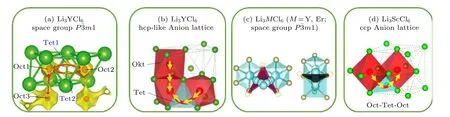
圖10 傳導離子在LTMH (Li3TMCl6)中輸運特性 (a) Li+在Li3YCl6 中輸運路徑[110]; (b) Li+在hcp 陰離子晶格Li3YCl6 中輸運路徑[111]; (c) Li+在Li3ErCl6 和Li3YCl6 中的輸運行為[112]; (d) Li+在ccp 陰離子晶格Li3ScCl6 中輸運路徑[113]Fig. 10.Transport characteristics of conductive ions in LTMH (Li3TMCl6): (a) The ion transport in Li3YCl6 with space group[110]; (b) the ion transport in Li3YCl6 with hcp-like Anion lattice[111]; (c) the ion transport in Li3MCl6 (M = Y, Er) with space group [112]; (d) the ion transport in Li3ScCl6 with ccp Anion lattice[113].
基于多離子協同輸運特性, Nazar 團隊[114]對過渡金屬鹵化鋰Li3MCl6(M= Y, Er)進行Zr 摻雜制得Li3—xM1—xZrxCl6固體電解質, 室溫離子電導率1.4 mS·cm—1, 且能夠在高電壓下穩定工作. Zr對M的部分進行置換使得晶體結構發生三斜到正交的相變, 而置換導致的新Li3 位點在改變體系能量分布中起著重要作用, 在此過程中Li+與Cl—產生弱相互作用而有利于離子電導率. 同時, 鋰位引入的空位也提高了離子電導率.
液態、聚合物以及無機固態電解質之間離子運輸形式由含輸運離子溶劑化分子在梯度場中運動逐漸演變為聚合物鏈段運動與配位傳遞共同輸運離子, 最后演化為具有一定剛性骨架通道傳導離子遷移(如圖11 所示). 離子運動形式從間接的被動輸運逐漸轉變為直接的主動遷移. 然而, 在無機固態電解質中多離子協同效應與溶劑分子或聚合物鏈段協調離子輸運有一定相似之處. 同時, 電解質中無序化現象也是離子輸運的主要影響因素, 輸運過程中與離子相互作用的介質由運動狀態逐漸變為靜止狀態, 相互作用的介質由分子變為配位再到骨架離子. 雖無機固態電解質實現骨架通道形式,但離子占位的無序性影響其離子電導率. 電解質的發展正在趨于通過離子輸運微觀物理圖像的準確描述來優化其中協同效應與有序無序之間的關系,進而演變出利于離子輸運的合理結構.
電解質中離子輸運微觀物理圖像的描述可清晰表達離子的動力學行為, 其對電池的循環性能至關重要. 2016 年, 以物理、材料和化學等交叉學科融合的視角, 我們系統總結了第一性原理計算、分子動力學、相場模擬、有限元分析和機器學習等多尺度計算方法應用于鋰離子電池材料及器件的研究現狀, 并指出實驗和計算手段相結合有利于全方位呈現電子/離子輸運的微觀物理圖像[117]. 對于電解質固有離子輸運特性已獲得較清晰的描述, 但是界面離子輸運較為復雜, 單一固態電解質中的晶界、復合固態電解質中的界面、以及電解質與電極間界面離子輸運特性均為重要研究問題.
針對復合固態電解質中離子輸運, 我們從各個時空尺度上剖析了復合固態電解質中相-相界面的離子傳輸機制, 提煉出了多尺度下鋰離子傳輸機制, 并搭建了復合固態電解質的跨尺度理論設計框架[8]. 對于電解質與電極間界相離子輸運的研究中,相對于液體電解質與電極間具有較好的潤濕性, 固態電解質與電極間固-固接觸界面離子輸運微觀物理圖像的描述更為復雜[118]. 采用實驗方法表征離子輸運圖像的技術較多, 如固態磁共振成像、核磁共振技術、掃描電鏡、透射電鏡、中子深度剖析技術、光學顯微鏡等. 在對固態電池中離子輸運過程中固-固界面結構和化學變化的圖像表征技術的總結中, Lou 等[119]重點分析了3D 電極與固態電解質間界面中孔隙率和彎曲度所描述的有效離子傳輸行為. 通過電極的三維重構準確獲取微觀參數,多維成像技術描述離子傳輸路徑.
4 總結與展望

圖11 液態、聚合物以及無機固態電解質離子運輸形式 (a) 液態電解質中溶劑分子協調離子輸運[115]; (b) 聚合物基電解質中鏈段運動與離子輸運[116]; (c) 具有骨架通道NASICON 中多離子協同輸運[23]Fig. 11. Transport form of ion in the liquid, organic polymer and inorganic solid electrolytes: (a) Li+ coordination in electrolyte[115];(b) Ion coordinated transport in the single-ion solid-state polymer electrolytes[116]; (c) Concerted migration of multi-ion in NASICON with framework channels[23].
為清晰勾勒電解質中離子輸運物理圖像, 本文系統分析了不同物理形態電解質相關的離子輸運理論模型、輸運機制以及輸運實踐案例. 從電解質的有序化程度審視, 無論是液態電解質還是柔性聚合物電解質均表現為在無序狀態的介質中輸運離子. 無機固態電解質雖晶體結構表現為有序物理空間構型, 但是離子輸運過程中骨架離子的占位又表現出了無序狀態. 因此各電解質中刻畫離子輸運圖像的本征因素均表現為無序傳導. 如: 在研究Zn 離子嵌入和脫出NaV2(PO4)3的過程中發現,Zn 離子的嵌入激活了M1 位置的Na 離子, Zn 離子在NaV2(PO4)3中傾向于與M1 位Na 離子協同遷移, 由此形成Na+/Zn2+混占現象[48].
由液態到固態, 在克服離子傳導率降低趨勢的過程中, 目標是在無機固態電解質中實現類似液態電解質中的輸運圖像. 從輸運模式的角度進行分析, 相較于電子的有序傳導和超導現象, 離子輸運未來的發展是否也會演化為有序傳導并實現離子超導值得深入探究. 而有序化程度并非離子輸運成像描述的唯一因子, 為此應充分利用眾多描述因子獲得成像機制. 并且從離子輸運物理圖像逆向出發, 深入研究并提出相應的傳導模型與輸運機制,進一步建立離子輸運物理圖像與離子傳導率的定量關系. 基于此, 我們已搭建由材料物化參數計算程序、高通量計算任務管理系統和材料數據庫組成的固體電解質高通量篩選平臺(SPSE). 該平臺由微觀結構幾何分析(CAVD)、鍵價和(BVSE)計算、融合幾何方法與鍵價和計算的離子輸運通道分析、多精度融合算法、離子輸運描述因子計算、結構匹配、熱力學相圖構建等組成[120-122], 并成功部署在國家超級計算廣州中心且已對外開放供科研人員使用。
目前, 基于SPSE 平臺已完成多種陽離子與陰離子無機化合物的輸運特性數據庫的搭建[22]. 該離子輸運圖像描述方法已在固體電解質Li3PS4和Li10GeP2S12晶體結構及其離子輸運特性的研究中較好應用[123,124]. SPSE 平臺通過快速預測遷移離子最小能量路徑與自動生成的過渡態來避免人工干預, 實現離子輸運高通量計算. 進一步, 通過深度剖析CAVD 與BVSE 離子輸運微觀物理圖像中所得移動離子濃度與位點數差異, 及遷移路徑是否涉及未被占用間隙位點等信息, 亦可預測基本的輸運機制. 對于復合電解質可采用基于有效介質理論(EMT)和Halpin-Tsai 模型的離子輸運微觀物理圖像刻畫方法, 耦合機械性能與離子輸運特性以及固體電解質的致密度等因素, 高效篩選復合固態聚合物電解質(CSE)[125], 進而改善其離子電導率. 由于復合固態電解質是由不同相組成的離子導體, 為此, 我們通過多尺度探索離子遷移動力學提煉出其離子輸運圖像, 以審視設計新型CSE 的一般策略[8].
在電解質的研究中, 通過調控離子輸運微觀物理圖像的描述因子來改善離子輸運特性必將成為設計具有較好性能的電解質的理論依據. 對于離子輸運微觀圖像起決定性作用的物理本質: 晶格動力學、空間電荷層等的挖掘, 以及多尺度離子輸運的配合關系與物理屬性均為極具挑戰的課題[126]. 此外, 離子輸運圖像也會受到外載荷作用的影響. 例如, 高壓會誘導材料的結構發生相變, 進而影響離子輸運的微觀物理圖像[127]. 目前來看, 使固態電解質的離子輸運特性達到液體電解質的水平是未來固態電解質的主要發展方向之一. 通過多尺度理論仿真計算電解質的電化學窗口穩定性和與正極材料的相容性[128], 以及分析離子輸運行為預測離子電導率[129], 或計算預測同二元系傳導性質相反的離子輸運特性[130,131]等均已成為該領域的重要途徑. 同時, 融合專家領域知識的數據驅動離子輸運微觀圖像研究, 也是未來的方向之一[132-134]. 電解質中離子輸運微觀物理圖像的清晰描繪為其合理匹配整個電池系統提供科學依據, 并為提高電池的整體性能奠定基礎.
