營養性肥胖大鼠模型腸道菌群的微生物多樣性分析
2020-12-09 03:41:06李宏睿張德琴金曉蕾王大文辛鵬飛何進程麻富斌周彩云孫曉瑋張建剛
中國比較醫學雜志 2020年11期
關鍵詞:物種
李宏睿,張德琴,金曉蕾,唐 驍,王大文,高 雪,辛鵬飛,何進程, 麻富斌,周彩云,孫曉瑋*,張建剛*
(1.蘭州大學基礎醫學院病理學研究所,蘭州 730000; 2.山丹縣人民醫院內科,甘肅 山丹 734100)
隨著生活水平的提高,超重和肥胖的人數迅速增加,肥胖癥已成為健康的主要危險因素之一。能量攝入過多是肥胖發生的主要原因,作為能量物質最終進入機體內環境(吸收)的門戶和屏障,小腸具有重要的研究價值[1]。越來越多的證據表明腸道菌群參與營養性肥胖的發生[2-5]。人的腸道內有大約10萬億個細菌,大多分布于結腸,由于樣本主要從糞便中獲得,目前大多數研究實際為結腸菌群研究[6-8],然而結腸糞菌是否能夠反映小腸的菌群特征,反映與吸收功能及營養性肥胖的確切關系,尚缺乏研究。結腸和小腸菌群差異是營養性肥胖發生的腸道菌群機制研究要解決的關鍵科學問題[9]。
本文基于前期建立的營養性肥胖大鼠模型,通過16S rRNA測序,對高脂膳食性大鼠模型回腸和結腸內菌群的微生物多樣性進行分析,以明確小腸和結腸菌群的分布特點及其與營養性肥胖發生的關系。
1 材料和方法
1.1 實驗動物
清潔級雄性Sprague-Dawley(SD)大鼠43只,4~5周齡,體重(52.32±8.48) g,購于蘭州大學實驗動物中心 [SCXK(甘)2018-0002],。按照實驗要求,為模擬自然狀態,所有動物在蘭州大學病理學研究所動物實驗室普通環境下單籠飼養[SYXK(甘)2018-0002],自由進食水。動物實驗符合國家實驗動物福利相關規定,并獲得蘭州大學醫學倫理委員會批準(jcyxy20190302),實驗過程嚴格按照《實驗動物管理與使用指南》進行,嚴格遵守動物使用的“3R”原則,實驗過程給予大鼠以人道主義關懷。
1.2 主要試劑與儀器
高脂飼料按照豬油(lard)∶蛋黃粉(egg yolk powder)∶基礎飼料(chow powder)∶蔗糖(sugar)=1∶1∶1.5∶0.4配制[10],能量5.58 kcal/g。豬油從市場購買,新鮮煉制;蛋黃粉購自北京金健力蛋粉廠(蛋白質31.6%,脂肪55.1%,碳水化合物5.3%,巴氏滅菌);SPF級基礎飼料(能量3.16 kcal/g)購自北京科澳協力有限公司,60Co 滅菌;蔗糖從市場購買。NucleoSpin 96 Soi DNA提取試劑盒,德國Macherey Nagel公司; KOD FX Neo DNA聚合酶,日本Toyobo公司;Phusion DNA聚合酶,美國NEB公司;DNA純化柱,美國Omega Biotek公司;Monarch DNA膠回收試劑盒,美國NEB公司。ZK-26/100型真空泵,杭州米歐儀器有限公司;SynergyHTX型酶標儀,美國Gene公司;高速離心機,美國Eppendorf公司;9902型PCR儀,美國Allen Bradley公司。
1.3 實驗方法
1.3.1 實驗分組及營養性肥胖大鼠模型建立
動物按體重隨機分為普通飼料組(chow group,n=10),高脂飼料組(high fat diet, HFD group,n=33),適應性喂養10 d后,進行混合喂養[10],高脂飼料組同時飼喂高脂飼料和普通飼料,普通組單純飼喂普通飼料,建立營養性肥胖模型。混合喂養60 d,分別以各組前15%為肥胖傾向(>對照組平均體重20%, obese prone, HFD-OP,n=5),以后15%為肥胖抵抗(obesity resistance, HFD-OR,n=5),再以普通飼料繼續喂養60 d,去除HFD食物本身誘導對腸道菌群的影響[11-13]。
1.3.2 腸道內容物收集
動物麻醉后皮膚消毒,無菌條件下開腹,門靜脈采血處死動物,提取回腸及結腸,分離內容物,并立即于冰上分裝至2 mL無菌EP管中,每管樣品量0.5~2.0 g,液氮速凍,-80℃保存備檢。
1.3.3 微生物多樣性分析
利用Illumina測序平臺(百邁客生物科技公司,北京),采用Illumina HiSeq 2500雙末端測序(paired-end)檢測微生物多樣性,測序長度350 bp~450 bp。
PCR擴增及測序文庫構建:提取菌群總DNA,根據保守區設計細菌16S rRNA基因V3+V4區引物,引物序列如下:338F:5′-ACTCCTACGGGAGG CAGCA-3′;806R:5′-GGACTACHVGGGTWTCTAAT-3′。引物末端加barcode,對目標區域PCR擴增,擴增程序如下:95℃ 5 min→95℃ 30 s→50℃ 30 s→72℃ 40 s 25 cycles→72℃ 7 min→4℃。擴增產物經純化、定量和均一化,形成測序文庫。
測序:PCR擴增產物質檢后制備flow cell芯片,Illumina Hiseq 2500雙端測序,得到原始圖像數據文件,經堿基識別(base calling)分析轉化為原始測序序列(sequenced reads)。
數據預處理:根據PE reads之間的overlap關系,將Illumina測序得到的雙端序列數據拼接(FLASH, v.1.2.11, JHU Center for Computational Biology)[14],將拼接得到的序列進行質量過濾(Trimmomatic, v.0.33, USADELLAB.org)[15],去除嵌合體(UCHIME, v.4.2, USEARCH 11)[16],成一條序列tags。
OTUs(operational taxonomic units)分析:對測序數據進行質量評估,使用Usearch軟件(v.10.0)[17]對tags在97%的相似度水平下進行聚類,獲得OTU,一個OTU代表一個物種。使用Mothur軟件(version v.1.30, http://mothur.org/),對各個樣品的alpha多樣性指數進行評估,使用QIIME軟件(v2.2, SCIKIT-BIO, http://qiime.org/)進行beta多樣性分析,比較不同樣品在物種多樣性方面存在的相似程度。
物種注釋及分類學分析:將OTU的代表序列與Silva微生物參考數據庫(v. 128, http://www.arb-silva.de)[18]進行比對,得到每個OTU對應的物種分類信息,應用RDP Classifier(v2.2, QIIME)[19]對OTU進行分類學注釋。進而在門、綱、目、科、屬、種(phylum, class, order, family, genus, species)各水平統計各樣品群落組成,利用QIIME軟件生成不同分類水平上的物種豐度表,R語言繪制群落結構圖。
1.4 統計學方法
2 結果
2.1 營養性肥胖模型
建模60 d,普通飼料組體重為(326.51±29.29) g,高脂飼料組體重為(355.99±36.33) g(P<0.05)。高脂飼料組15%個體體重(HFD-OP)> 對照組平均體重20%(5/33),與普通飼料組及相應15%低體重(HFD-OR)(5/33)相比,差異具有統計學意義(P< 0.05),HFD-OR與普通飼料組相比,差異無統計學意義(P>0.05)(表1)。
24 h初始進食量普通飼料組為(9.23±2.01) g,高脂飼料組為(9.10±1.71) g,差異無統計學意義,P>0.05。建模期間,普通飼料組進食量為(980.00±53.43) g(普通飼料),高脂飼料組進行混合喂養,其中高脂飼料的進食量為(630.70±90.61) g,普通飼料的進食量為(337.88±82.34) g,高脂飼料與普通飼料的比值為(2.02±0.72)∶1,提示實驗動物更偏好高脂飼料。
HFD-OP大鼠在高脂飼料、普通飼料的進食量以及對食物的偏好與HFD-OR大鼠無顯著性差異(P>0.05)(表1)。
2.2 低能量膳食對營養性肥胖的影響
經普通飼料繼續喂養60 d,OP大鼠的體重仍然顯著高于OR與普通飼料組,其腎周及睪周脂肪質量也顯著高于其它組,P<0.05,見表2。
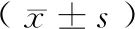
表1 普通飼料組與高脂飼料組大鼠體重及攝食情況Table 1 Body weight and food intake of rats in chow group and HFD group
2.3 腸道菌群多樣性分析
提取腸內容物總DNA,樣品質量符合測序要求,利用Illumina測序平臺對樣品DNA進行擴增、測序,得到2489179個PE reads,經質量過濾后共得到2208772個clean tags。各樣品GC含量為(54.21±0.97)%,Q30達到(93.96±1.61)%。測序長度在406~420 bp之間。
OTU分析:OTU數反映菌群的多樣性。使用Usearch軟件對tags在97%的相似度水平下進行聚類、獲得OTU。經過60 d的飼料均一化干預,高脂飼料組回腸OTU數與普通飼料組無明顯差異,但結腸OTU數顯著降低,表明高脂膳食引起大鼠結腸微生物豐度下降,見表3。
不同體重回腸與結腸OTU數無顯著性差異,見表4。
Alpha多樣性分析回腸、結腸菌群的OTU,獲得ace、chao1、simpson、shannon指數,多元多因素方差分析顯示,體重、飼料、部位因素之間無交互作用(P>0.05,數據未提供),飼料與體重對alpha多樣性指標無明顯影響(P>0.05,數據未提供),但回腸與結腸間alpha多樣性差異具有統計學意義,回腸菌群的chao1、ace指數均顯著高于結腸,表明回腸菌群中細菌數量(豐度)遠高于結腸,而多樣性低于結腸(simpson、shannon)(P<0.05)(表5)。Venn圖顯示,除普通飼料回腸外,其余組內各樣品間OTU具有較大的重疊性,圖1。
2.4 回腸與結腸菌群beta多樣性分析
應用非加權Binary_Jaccard距離算法比較不同樣品在物種多樣性方面存在的相似程度,UPGMA聚類樹圖顯示樣品間菌群結構相似,但回腸與結腸樣品間的相似度較低(圖2)。
表2 低能量膳食對營養性肥胖大鼠體重及內臟脂肪的影響Table 2 Effects of low energy diet on body weight and visceral fat of nutritional obese rats
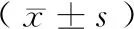
表3 普通飼料組及高脂飼料組回腸、結腸菌群OTU數比較Table 3 Comparison of OTUs in ileum and colon of chow group and HFD group
表4 不同體重大鼠回腸、結腸菌群OTU數比較Table 4 Comparison of OTUs in ileum and colon of rats with different weights

注:除普通飼料回腸,各組個體間共有的OTU數顯著高于個體特有的OTU數。圖1 回腸、結腸細菌OTU的Venn圖Note. Except for chow-ileum, the number of Common OTUs in each group was significantly higher than that of individual’s specific OTUs.Figure 1 Venn diagram of OTU in ileum and colon
表5 回腸、結腸菌群alpha多樣性指數比較
進一步采用非度量多維標度(nonmetric multidimensional scaling, NMDS)、主坐標分析(principal coordinate analysis, PCoA)和主成分分析(principal component analysis, PCA),對腸道微生物群落結構的變化進行分析,如圖3所示,結腸樣本間菌群組成顯示出明顯的相似性,而回腸與結腸以及回腸樣本間相似度較低。
2.5 物種注釋及分類學分析
將OTU的代表序列與微生物參考數據庫Silva進行比對,得到每個OTU對應的物種分類信息。結果顯示,在屬水平上,回腸與結腸優勢菌(豐度排名top 10)存在比較大的差異,回腸內的主要優勢菌為Romboutsia、Turicibacter、Rothia、Lachnospiraceae、Streptococcus、Candidatus_Arthromitus、Helicobacter、Lactobacillus等,而結腸的主要優勢菌為Lachnospiraceae、Treponema、Akkermansia、Candidatus_Saccharimonas、Ruminococcaceae、Romboutsia等,兩者優勢菌的重疊程度較低,見圖4、圖5。
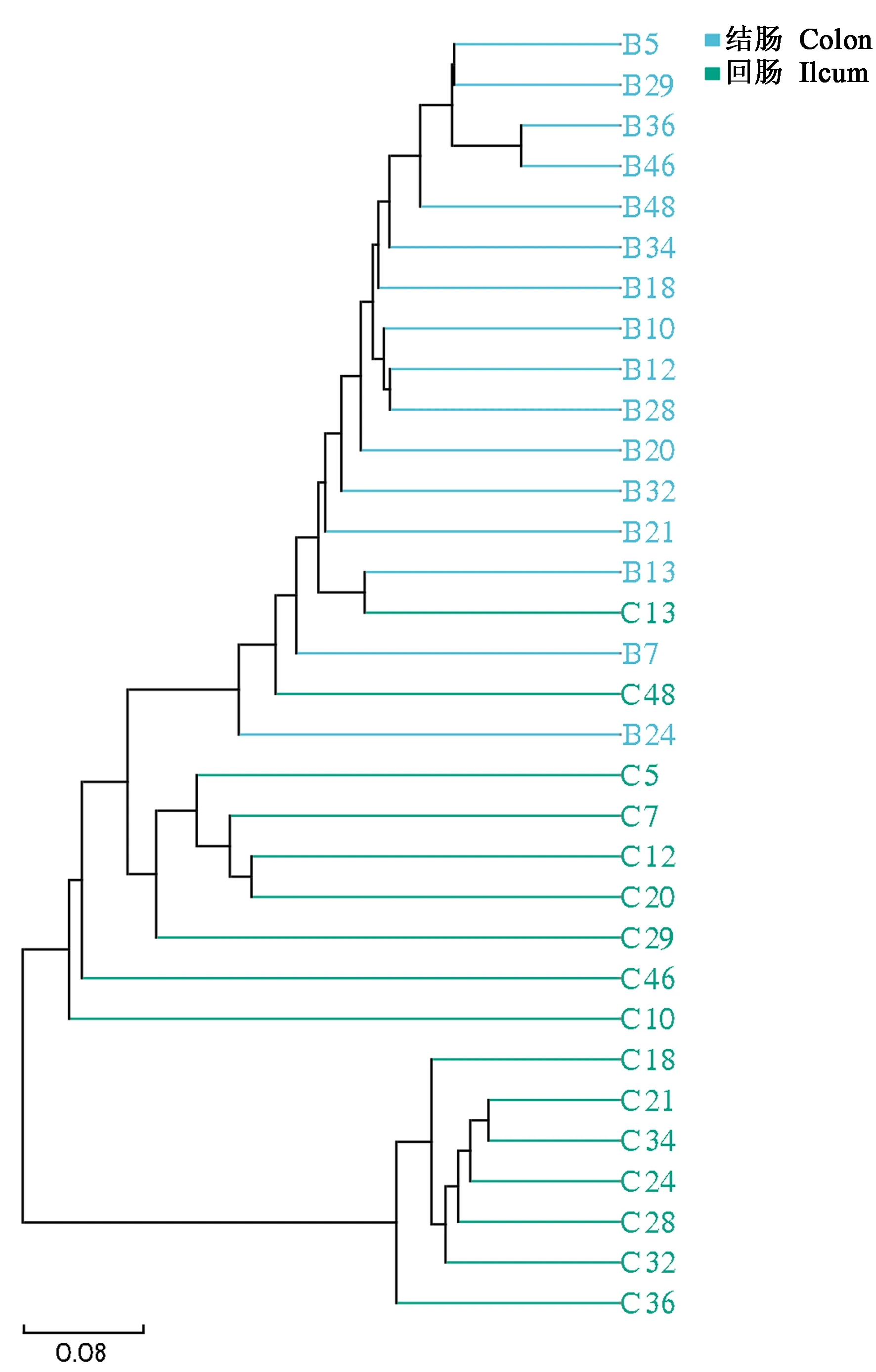
注:B:結腸;C:回腸。圖2 UPGMA聚類樹圖顯示回腸、結腸細菌 結構特征Note. B, Colon. C, Ileum.Figure 2 UPGMA cluster tree showed the structural characteristics of ileum and colon microbiota
HFD大鼠回腸Turicibacter低于普通飼料組,而結腸內的Treponema、Candidatus_saccharimonas低于普通飼料組,證明高脂膳食對回腸和結腸菌群的影響不同,并且這種影響具有持久性。
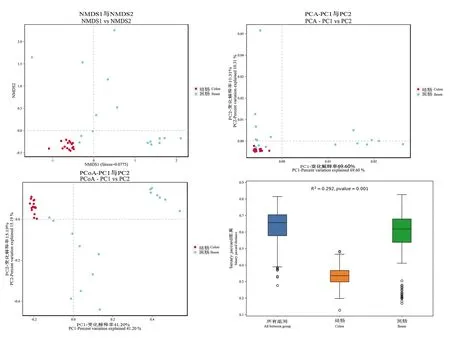
圖3 NMDS、PCA、PCoA分析法顯示回腸、結腸細菌結構特征Figure 3 NMDS, PCA and PCoA analyses showed the structural characteristics of ileum and colon microbiota

圖4 回腸內細菌相對豐度和物種注釋(屬水平)Figure 4 Relative abundance and bacteria annotation in ileum (genus level)

圖5 結腸內菌群相對豐度和物種注釋(屬水平)Figure 5 Relative abundance and bacteria annotation in colon (genus level)

注:B:結腸;C:回腸。圖6 HFD-OP回腸、結腸內細菌物種豐度聚類熱圖(屬水平)Note. B, Colon. C, Ileum.Figure 6 The cluster heat map of bacterial species abundance in the ileum and colon of HFD-OP rats (genus level)
回腸是營養物質吸收的主要部位,HFD-OR大鼠回腸內Rothia菌豐度明顯增加,而Romboutsia菌豐度降低,提示Rothia菌和Romboutsia菌可能參與回腸吸收的調節作用。此外,HFD-OR結腸內Ruminococcaceae_UCG-005顯著增加。
聚類分析熱圖顯示HFD-OP大鼠回腸與結腸內菌群類型的差異,HFD-OP大鼠回腸內Lachnospiraceae、Candidatus_Arthromitus、Helicobacter、Lactobacillus較其它處理組豐度低,而Turicibacter、Romboutsia、Streptococcus、Rothia的豐度較高。與其它各組相比,結腸內菌群除與其它各組相同的優勢菌群外,Prevotella_9、Prevotellaceae_Ga6A1_group豐度較高,而Candidatus_Saccharimonas、Lactobacillus、uncultured_bacterium_f_Ruminococcaceae豐度較低,圖6。
3 討論
營養性肥胖是一種嚴重威脅人類健康的全球公共衛生問題,腸道作為能量物質最終進入機體內環境的門戶和屏障,其內大量定居的菌群被認為在肥胖的發生中發揮著重要作用[20]。腸道菌群作為機體重要的共生生命,與食物成分及消化道的功能相互協同,形成一個微生態系統,在消化、營養、免疫等方面發揮重要生理功能,若其構成改變也會導致心、腦等器官發生多種嚴重疾病[21-23]。哺乳動物腸道微生物數量存在縱向變化,例如在人的消化道內,細菌數量呈指數級增長[24],胃、小腸近端、回腸、結腸中每毫升內容物的細菌數量分別為102~103、103~105、108、1011。結腸是機體最大的食物殘渣分解場所,長期以來,腸道菌群的研究主要集中在結腸,那么結腸糞菌能否代表小腸菌群?本研究表明,盡管回腸與結腸之間菌落的OTU數差異不明顯,并且多數菌群存在重疊(Venn圖),但alpha多樣性分析顯示回腸菌群的樣品內物種豐度顯著高于結腸(chao1、ace),而多樣性低于結腸(simpson、shannon);beta多樣性分析顯示結腸樣本間菌群組成具有明顯的相似性,而回腸與結腸以及回腸樣本間相似度較低;經注釋分析,兩者的優勢菌群存在明顯的不同,并且重疊程度較低;聚類分析熱圖也顯示回腸與結腸內的菌群物種豐度分布不同,表明結腸內的糞菌并不能代表回腸內的菌群結構。因此,在腸道菌群研究中必須注意這一問題,這與Leite等[25]對人、Grond等[26]對濱鳥、Su等[27]對蒙古馬的研究結果相一致。
小腸是營養物質吸收的主要場所,研究結論顯示,腸道菌群通過多種機制參與肥胖的發生,包括LPS誘導的腸滲漏和炎癥[28],慢性低度內毒素[29]、腸源性肽激素[30]、脂肪組織活性成分等的調節[31],包括GPCR、AMPK信號途徑影響能量平衡和飽食感,以及短鏈脂肪酸影響脂類和能量物質代謝[32]等,但多數研究是基于結腸糞菌探討與肥胖的關系,還不足以揭示小腸菌群調節吸收促進營養性肥胖發生的機制。我們的研究發現,回腸內的主要優勢菌為Romboutsia、Turicibacter、Rothia、Lachnospiraceae、Streptococcus、Candidatus_Arthromitus、Helicobacter、Lactobacillus等,而結腸的主要優勢菌為Lachnospiraceae、Treponema、Akkermansia、Candidatus_Saccharimonas、Ruminococcaceae、Romboutsia等,提示回腸與結腸菌群存在功能的差異,小腸菌群和糞菌可能具有不同的功能屬性。
OTU是一個相對穩定的指標,不容易受到營養程度的影響,但本研究表明,高脂膳食可能對結腸的OTU數存在比較持久的影響,物種注釋顯示高脂膳食對回腸、結腸內的優勢菌也具有持久的影響。我們發現,Turicibacter、Rothia、Romboutsia、Streptococcus可能參與回腸吸收的調節,在營養性肥胖的發生中發揮一定的作用,對于這一結果,還需要更多的動物實驗研究支持。
腸道菌群研究是一個非常復雜的領域,一方面由于腸道微生物組成在個體間具有很大的差異,而且不同背景(飲食習慣和種族)的參與者構成及用于分析細菌的不同方法在很大程度上都會影響研究結果的統一;另一方面則因為疾病動物模型在腸道微生物群組成、發酵過程、進食習慣等方面存在差異[33]。本文在營養性肥胖大鼠模型的基礎上,研究腸道菌群與肥胖發生的關系,進一步證實結腸與回腸具有不同的菌群特征,對于探尋人類腸道菌群促進營養性肥胖發生的機制具有較好的借鑒價值。
綜上所述,本研究通過對高脂膳食性營養性肥胖大鼠回腸和結腸菌群進行16S rRNA基因測序分析,證實回腸與結腸具有不同的腸道菌群,結腸菌群并不能代表回腸的菌群特征,腸道的部位是進行腸道菌群研究必須要考慮的因素;Turicibacter菌、Rothia菌、Romboutsia菌、Streptococcus菌可能是參與肥胖發生的關鍵回腸固有菌群。
猜你喜歡
課堂內外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2024年12期)2024-12-02 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
中學生博覽(2022年7期)2022-06-21 21:48:14
大科技·百科新說(2021年8期)2021-11-03 10:55:16
學苑創造·A版(2021年5期)2021-06-28 19:51:42
少兒美術(快樂歷史地理)(2020年9期)2020-03-19 05:10:56
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
汽車觀察(2018年10期)2018-11-06 07:05:26
