Angelman 綜合征家系的臨床和遺傳學特點
2020-12-15 07:03:20程書歡李蒙蒙程亞穎
吉林大學學報(醫學版) 2020年6期
程書歡, 孫 萌, 李蒙蒙, 程亞穎
(河北省人民醫院兒科,河北 石家莊 050051)
Angelman 綜合征(Angelman syndrome, AS)是由于母源性泛素蛋白連接酶E3 (ubiquitinprotein ligase E3A, UBE3A) 基因功能缺陷而致的神經發育障礙性疾病。 發病率約為1/150 000,其中大約70% 是由于UBE3A 基因所在區域15q11~q13 的缺失所致,10% 是由于UBE3A 基因致病變異所致。現國內外報道的UBE3A 基因突變以移碼突變為主,且多為個案報道,家系報道的病例極少。本研究對2019 年3 月河北省人民醫院確診的AS 家系進行回顧性分析,本家系中兄弟二人均發病,UBE3A 基因突變為無義突變,為未報道過的新突變,這豐富了UBE3A 基因突變譜。本文作者將該家系的臨床特征和基因遺傳學特點進行總結,以提高臨床醫生對AS 的認識。
1 資料與方法
1.1一般資料患兒,男性,出生4 個月3 天,因“發育落后1 個月余” 于2018 年11 月14 日就診于河北省人民醫院兒科。 患兒系37+4周, 孕4 產3(G4P3),因其母胎膜早破經陰道分娩娩出,羊水色清,臍帶扭轉20 周,胎盤未見明顯異常,Apgar評分均為10 分。在孕32 周時出現宮縮,曾保胎治療(具體用藥情況不詳)。胎兒出生體質量3.2 kg,出生時頭圍和身長不詳。母乳喂養,生后數天開始頻繁吐奶,2 個月后有所緩解。父母體健,非近親結婚。其母孕期無特殊用藥史,無污染和放射線接觸史。否認家族性及遺傳性疾病史。查體: 體質量6.5 kg,身長65 cm,頭圍40.8 cm,神志清楚。雙眼可注視,不能追視。很少咿呀發音,逗之可微笑,不能笑出聲。不能豎頭,俯臥位頭短暫抬離床面1~2 s,胸不能抬離床面,肘不能支撐。不會玩手,雙手不能握持玩具,不會伸手取物,不會翻身。被動輔助站位時雙腳尖著地。前囟平坦,頸無抵抗,心肺腹查體未見明顯異常。雙側髖關節外展稍受限,四肢肌張力高。患兒時有左上肢屈曲上舉至面部、雙下肢并攏屈曲上抬,持續約10 s 緩解,每日數次發作,多在平臥位清醒時發作,行為刻板,發作時雙眼無凝視,無顏面和口唇發紺。奶后偶有嘔吐。輔助檢查:肝腎功能、電解質和甲狀腺功能五項未見異常。頭顱MRI 未見明顯異常。視頻蝶骨腦電圖:間歇期,各導聯少量彌漫性慢波,后頭部夾雜棘波;監測到1 次清醒期發作,表現為左上肢上舉,雙下肢屈曲伴有頭部的不自主抖動,持 續 約 1 min 緩 解; 而 同 期 腦 電 圖(electroencephalogrphy, EEG) 可 見 大 量 動 作 偽跡,未見癲癇樣放電,提示非癲癇發作。血尿代謝篩查無明顯異常。詳細詢問家族史,得知患兒的二哥, 年 齡5 歲2 個 月, 系36 周 早 產 兒, 孕3 產2(G3P2),經陰道分娩娩出,羊水、臍帶、胎盤及Apgar 評分不詳。 嬰兒期因患兒易吐奶, 經常患“吸入性肺炎”。生后發育遲緩,1 歲左右會獨坐,1.5 歲會爬,近2 歲會說話、獨走,步態不穩。1 歲內睡眠差,很難哄睡。自4 歲7 個月開始出現癲癇發作,表現形式多樣,有時表現為玩耍過程中突然跌倒,意識喪失,雙眼緊閉,頭后仰,四肢軟,無口周發紺、牙關緊閉,持續約1 min 緩解,緩解后精神反應如常;有時僅表現為右側上肢抖動;有時表現為進食時突然停止,閉目,手中的東西掉落,呼之不應,持續3~5 s 后緩解。有時1 d 內間斷發作十余次。患兒只能敘述單個字,不會說疊音字及單詞,不會說3 個字以上的短句,可用簡單手勢及表情表達意愿,有時能聽懂簡單的指令。查體:體質量20 kg, 身高110.4 cm, 頭圍52 cm。 神志清楚,皮膚和頭發顏色無異常。常無原因的笑,容易興奮,多動。行走時喜上舉上肢,左右搖擺走路,步態不穩。嘴較大,牙縫寬,牙釉質發育差,右上尖牙先天性缺失。心肺腹查體未見異常。四肢肌力和肌張力尚可,雙側腱反射亢進,病理征均陰性。輔助檢查:肝腎功能、電解質、血氨、血同型半胱氨酸和甲狀腺功能六項無異常。頭顱MRI 平掃未見明顯異常。 視頻蝶骨EEG (圖1, 見封三) 顯示:雙側枕和后顳區可見大量中高幅棘慢波、慢波同步或非同步發放;雙側半球可見大量彌漫性中高幅2.5~3.5 Hz 短至中程慢波陣發性發放,雙側額區可見棘慢波發放,血尿代謝篩查無異常。患兒的大哥,年齡10 歲9 個月,出生史無異常,智力正常,體格發育正常。小學四年級,學習成績尚可。
1.2基因檢測取得患兒家長同意后,分別抽取兄弟二人及其父母外周血2 mL 送檢北京邁基諾醫學檢驗所進行基因分析,使用基因組DNA 提取試劑盒提取DNA,純化擴增后采用高通量測序系統Nextseq500 (Illumina 公司) 對4 450 個臨床相關基因的外顯子區域進行二代基因測序,陽性檢測結果采用Sanger 測序方法進行父母驗證;并采用Sanger測序方法對表型正常的其他家庭成員(患兒的大哥、患兒的舅舅和患兒的姨母) 進行檢測。采用SIFT、 PolyPhen_2 和REVEL 等多種生物學軟件對突變位點進行生物信息學致病性預測。按照美國醫學遺傳學與基因組學學會(American College of Medical Genetics and Genomics, ACMG) 發 布 的最新基因突變解讀標準和指南[1]對相關基因進行總結,分析是否存在與疾病表型相關的致病性突變。二代基因測序結果顯示:患兒及其二哥均存在1 個UBE3A 基因雜合突變位點chr15-25616495,導致第4 外顯子編碼區第766 號核苷酸由胞嘧啶突變為胸腺嘧啶,導致氨基酸終止編碼(Cordially.766 C>T p.R256X)。患兒及其二哥的突變均遺傳自母親(圖2 A~D,見封三)。患兒大哥、舅舅和姨母該位點未見相關突變(圖2 E~G,見封三)。根據臨床表現和基因檢測結果繪制AS 患者家系圖(圖3)。
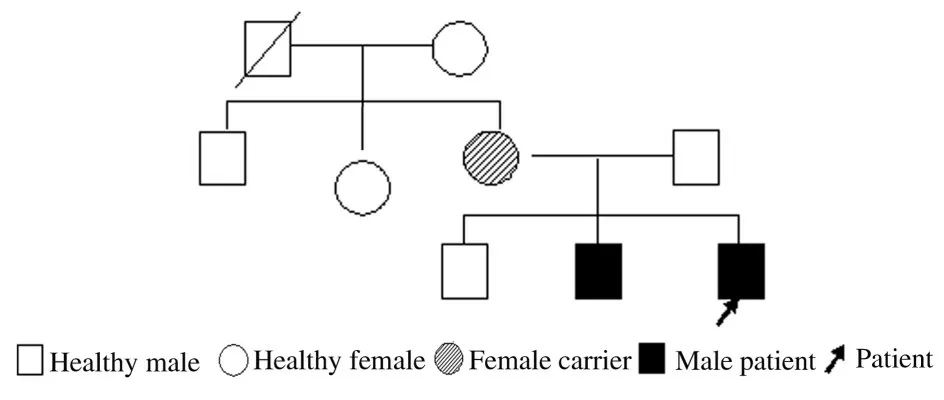
圖3 該AS 患者家系圖Fig.3 Familial pedigree of this AS patient
1.3治療和隨訪患兒確診后給予運動和認知康復訓練并隨訪,患兒經康復訓練后神經功能有所改善,7 個月時雙眼可追物,能喃喃發出咿呀發音,不能分辨家人。俯臥位可抬頭,雙肘支撐前胸能短暫抬離床面,手能握持玩具,不能伸手抓物。四肢肌張力較之前好轉。9 個月大時能單側翻身,能獨坐片刻,俯臥位肘支撐差,不會爬,被動輔助站位時雙足著地,喜媽媽抱。仍有左上肢上舉的刻板行為,無明顯癲癇發作。患兒的二哥給予口服丙戊酸鈉,至今無驚厥發作。患兒二哥與患兒同時進行語言康復訓練半年,其詞匯量較之前增多, 會認26 個單字及少許疊字,運動平衡能力有所提高。
2 討 論
AS 患者的主要臨床特征為神經發育遲滯,包括智力低下、運動障礙、語言落后、異常步態、快樂行為、 小頭畸形、 多動、 癲癇和特征性異常EEG 等[2], 其 致 病 基 因UBE3A 位 于 染 色 體15q11~q13 區域,該基因在正常腦組織中僅有母本基因優先表達,而遺傳自父系染色體的UBE3A 基因被特殊印跡而表達沉默。 導致AS 的遺傳學機制:由母源性15q11~q13 染色體缺失、父源性單親二倍體(uniparental disomy,UPD) 和印跡基因缺失等導致的15q11~q13 區域甲基化異常[3-4];母源UBE3A 基 因 存 在 致 病 性 突 變。 研 究[5]顯 示:AS 患者UBE3A 基因相關的突變有150 余種,突變形式多樣,其中60%~70% 為移碼突變導致的小缺失和重排,約25% 為錯義和無義突變,其他為剪接缺陷和復雜重排等。無義突變相對來說較少見,SADIKOVIC 等[6]總 結 了 近150 個UBE3A 基因突變患者,其中約35 個患者的UBE3A 基因發生了無義突變。本研究報道的該家系患兒UBE3A 基因中有1 個雜合突變, 導致第4 外顯子編碼區第766 號核苷酸由胞嘧啶突變為胸腺嘧啶,導致氨基酸終止編碼(c.766C>T p.R256X), 為無義突變。 ACMG 指南[1]顯示:該變異初步判定為疑似致病性突變,可能導致基因功能喪失,在正常人群數據庫中為低頻變異。經家系驗證分析:患兒母親該位點雜合突變,患兒父親、大哥及其他家庭成員該位點無突變。該家系中有兩兄弟均患病,且均攜帶相同的UBE3A 突變基因,因此并非患兒自身的新發突變,進一步發現其母親也攜帶該突變,證實該突變是由臨床表型正常的母親傳遞給患兒。經檢索NCBI ClinVar、HGMD 和PubMed 數據庫可見:這些數據庫中均無相同的基因突變。 因此,UBE3A 基因的c.766C>T 突變為未報道過的新突變, 豐富了UBE3A 基因突變譜。 患兒母親攜帶UBE3A 變異基因,但其臨床表型正常,患兒舅舅和姨母基因檢測未見異常,分析其母基因突變來源:一是由其臨床表型正常的父親傳遞,二是其父源性染色體特定區域發生了自身的新發突變。由于患兒外祖父已離世,故未能完善相關的基因檢測。
AS 所有遺傳機制均會導致患者出現一致的臨床特點:重度到極重度的智力低下、運動障礙、特征行為和嚴重發音及語言受限。 文獻[7-8]報道:AS 患者的臨床表型與基因型有一定的關系,攜帶染色體15q11~q13 區域大片段缺失的患者臨床表型明顯較其他基因型嚴重,表現為小頭畸形、運動障礙(如共濟失調、肌張力低下、喂養困難)、語言障礙、癲癇發作和皮膚色素減退等,缺失片段越大其臨床表現就越嚴重。對于缺失型來講,缺失片段越長,距離著絲點越近,語言損害、運動障礙和癲癇可能越嚴重。UPD 和印跡基因缺失的患者通常臨床表型相對較輕,小頭畸形、癲癇和色素減退發生率低,且共濟失調較輕和認知能力較高[9]。研究[9-11]顯示:攜帶UBE3A 基因突變患者的臨床表型最輕,該基因型患兒發育商最高,適應行為接近正常兒童。提前出現終止密碼子的患者臨床表型通常較重,該家系患兒臨床表型重,與既往研究[11]結論一致。
在AS 患者中癲癇發生率較高,且常在3 歲之前發生[12]。根據2005 年更新的AS 診斷標準, 癲癇發作的發生率為80%,通常發作嚴重且為藥物難 治 性。 沈 金 梅 等[13]研 究103 例AS 兒 童 發 現:有77.7% 患兒伴有癲癇發作,其中80.8% 患兒在3 歲以前出現癲癇發作。同時其特異性EEG 對診斷AS 也 具 有 重 要 意 義。 AS 患 者EEG 特 點[14-16]:①額區廣泛的高波幅2~3 Hz、200~500 μV 的δ 節律或三相δ 波;②后頭部為主的4~6 Hz、200 μV以上的θ 節律;③棘慢復合波多位于枕區,也可為廣泛性棘慢復合波發放,閉目時更容易誘發;④非進展性腦病的肌陣攣持續狀態,發作期的EEG 為彌漫性慢波短-長程發放,伴有肌陣攣、不典型失神和失張力等;⑤常用睡眠期的電持續狀態。患兒腦電圖可見彌漫慢波,后頭部夾雜棘波,較符合AS 患者EEG 背景活動。目前患兒年齡小,EEG 尚無癇樣放電,臨床亦無癲癇發作,需密切觀察。患兒二哥有癲癇發作,且EEG 檢測呈現典型AS 的EEG 特征。 因此, 對于臨床表現不典型的AS 患者,可早期行視頻EEG,依據其特異性EEG 結果協助診治。
各基因型AS 患者均有語言發育的缺陷,絕大多數患者語言完全缺乏,一小部分會使用少量單個字及疊音字,極少數患者可以使用短語。基因缺失型患者語言功能基本完全喪失,非缺失型患者通常會途敘述約20 字[5]。但語言發育障礙的AS 患者擁有廣泛的非語言交際行為,主要以手勢為特征[17]。運動障礙亦是AS 患者臨床特點之一。研究[18]顯示:UBE3A 基因表達異常或缺陷可能降低小腦蒲肯野細胞功能活性,從而導致小腦性共濟失調,引起運動功能障礙。同時,AS 患者的飲食和睡眠問題也很多見。多有嬰兒期的喂養困難,易發生吸入性肺炎。AS 兒童中較多睡眠效率差,有嚴重的夜間醒來問題,導致睡眠需求減少和異常的睡眠-覺醒周期[19]。
除語言方面,非缺失型患者較缺失型患者在其他各方面表現更佳。因此建議進行語言訓練時應重視非語言溝通方式,盡早使用輔助交流工具,如圖片卡或交流板。對AS 患者來說,早期的康復訓練可以提高患兒的認知和運動能力。
本家系中患兒年齡較小,臨床表現不典型。但通過追問其家系,發現其二哥的臨床表現較為典型。本家系中兩兄弟通過二代基因測序檢測有關致病基因突變位點得以確診。經過康復訓練后其認知和語言水平有所提高。因此對于臨床表現不典型的早期疑似病例應及時進行基因相關檢查,為進一步遺傳咨詢提供依據。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國民間療法(2021年5期)2021-06-09 09:21:04
中國生殖健康(2020年2期)2021-01-18 02:51:26
文苑(2020年4期)2020-05-30 12:35:30
小學生導刊(2018年13期)2018-06-29 03:49:00
小學生作文(中高年級適用)(2018年3期)2018-04-18 01:24:47
飲食科學(2017年5期)2017-05-20 17:11:53
華北電力大學學報(社會科學版)(2016年4期)2016-12-01 03:59:30
少兒科學周刊·少年版(2015年4期)2015-07-07 21:11:17
西南軍醫(2015年4期)2015-01-23 01:19:30
