放射性同位素碳-14標記毒死蜱的合成與分析
2020-12-18 08:03:40余凱翔楊征敏徐凌鋒李書琰陳滬飛
同位素 2020年6期
余凱翔,楊征敏,周 兵,徐凌鋒,李書琰,陳滬飛,王 偉
(1.浙江大學 原子核農業科學研究所,杭州 310058;2.上海啟甄同位素標記合成研究中心,上海 201403)
毒死蜱(Chlorpyrifos)是美國陶氏化學于1965年成功開發并商品化的乙酰膽堿酯酶抑制劑,屬硫代磷酸酯類非內吸性廣譜殺蟲殺螨劑。它具有觸殺、胃毒和熏蒸作用,能有效防治多種作物的地上和地下害蟲,是一種具有中等毒性、低殘留的農藥品種。盡管毒死蜱的專利早已過期,但因其具有其廣譜、高效等優點,因而毒死蜱仍然是世界殺蟲劑市場的主流品種之一。2007年,我國全面禁止使用5種高毒有機磷殺蟲劑,毒死蜱迅速搶占這些禁用品種留下的市場空間。迄今,毒死蜱依然是我國應用最廣泛的有機磷類殺蟲殺螨劑之一[1]。
伴隨著毒死蜱長期持續、大量使用而產生的慢性毒性、生態環境等與人體健康密切相關的系列問題引起了國內外學者和多國政府部門的關注,人們對于該殺蟲劑可能引起的潛在危害而擔憂[2-5]。放射性同位素示蹤法是深入開展上述問題研究進而評判毒死蜱安全性的常用方法之一[6-13],而放射性同位素碳-14標記毒死蜱是示蹤研究中理想的示蹤劑。從毒死蜱的化學穩定性、代謝穩定性、毒理學重要性和示蹤試驗的要求等角度考慮,具有芳香性的吡啶環是毒死蜱分子中理想的標記位置。文獻報道了一種吡啶環碳-14標記毒死蜱(O,O-二乙基-O-(3,5,6-三氯-2-[2,6-14C2]吡啶基)硫代磷酸酯),但該標記物的制備條件苛刻,須使用劇毒的[14C]氰化鉀和氯氣,且氯氣必須在高溫高壓(高壓釜)下反應,因而實驗操作具有較大的潛在危險性[14]。為克服該法不足,筆者設計并制備了一種替代該標記物的吡啶環碳-14標記毒死蜱(1,O,O-二乙基-O-(3,5,6-三氯-2-[2,6-14C]吡啶基)硫代磷酸酯[15]),標記物(1)合成的條件溫和,操作安全、可行性高。
1 儀器與試劑
1.1 主要儀器
本研究中放射合成工作在上海啟甄同位素標記合成研究中心完成;Varian 400 MHz核磁共振儀(以TMS為內標):美國瓦里安公司;Mini-Scan TLC薄層放射性掃描儀:美國BioScan公司;FSA:美國Aim Research公司;Waters Alliance e2695 HPLC-Acquity Qda MS/2489 UV高效液相色譜-紫外光譜/質譜聯用系統和Waters 2545 HPLC-2998 PDA制備型液相色譜系統(配備Waters fraction collector Ⅲ):美國Waters公司;Agilent 7890B GC-5977B MS氣相色譜-質譜聯用系統:美國Agilent公司;Tri-Carb 4910TR液體閃爍測量儀:美國PE公司;Typhoon FLA9500 IP多功能激光成像儀:美國GE公司;Milli-Q Reference S. Kit(18.2 MΩ/cm,25 ℃)超純水制備系統:默克化工技術(上海)有限公司;Sartorius BSA22 4S-CW(1 mg)和BT25S(0.01 mg)電子天平:德國賽多利斯公司。
1.2 主要試劑
[14C]碳酸鋇(比活度55.3 mCi/mmol,放化純度99.9%,化學純度98%):美國ARC公司;2,5-二苯基噁唑(PPO):高效液相色譜(HPLC)純度大于99.0%,日本TCI公司;1,4-雙(5-苯基-2-噁唑)苯(POPOP):HPLC純度大于99.0%,日本TCI公司;Optiphase HiSafe 3閃爍液:美國PE公司;HPLC和HPLC-MS用甲醇:分別為色譜級和質譜級,美國Fisher Scientific公司;其他試劑均為市售分析純,試劑按照文獻[16]方法純化。
2 實驗方法
毒死蜱(Chlorpyrifos)化學結構圖示于圖1,放射性同位素碳-14標記毒死蜱(1)的合成路線示于圖2。
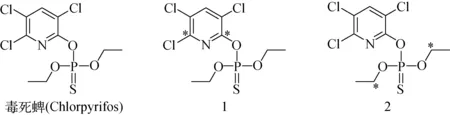
圖1 毒死蜱和兩種碳-14標記毒死蜱(1和2;*標示碳-14標記位點)Fig.1 Chlorpyrifos and its carbon-14 labelled counterparts (1&2, *indicates carbon-14 labelled sites)

圖2 放射性同位素碳-14標記毒死蜱(1)的合成路線(*標示碳-14標記位點)Fig.2 Synthetic route of radioisotope carbon-14 labelled chlorpyrifos (1, *indicates carbon-14 labelled sites)
2.1 放射性同位素標記化合物的制備
2.1.15-芐氧基[羰基-14C]戊酸(5) 利用自制的第五代微型集成式放射性二氧化碳反應裝置制備標記物(5)[7,17]。投料量和反應條件:(A)制備格氏試劑(4):依次向二氧化碳反應器中加入表面光亮的鎂屑(41 mg)、4-溴丁基芐基醚(3)的無水四氫呋喃(THF)溶液(5.1 mL,0.65 mol/L)和碘的無水THF溶液(0.5 mL,0.2 mol/L)進行反應。反應引發后控制加料過程中反應溫度為40~50 ℃;加畢室溫攪拌2 h。(B)格氏反應:在二氧化碳發生器中分別加入[14C]碳酸鋇(291.2 mg,55.3 mCi/mmol)和脫氣濃硫酸(4 mL);在-10 ℃進行格氏反應。按照標準操作程序啟動微型集成式反應裝置進行反應[7,17]。GC-MS和氣體放射性檢測表明,反應裝置運行2 h后[14C]CO2徹底消耗。向二氧化碳反應器中反應液里加入飽和氯化銨溶液(30 mL),減壓脫除該混合液中THF,殘余液以稀鹽酸(1 mol/L,下同)酸化(pH 2~3),二氯甲烷(DCM)萃取(35 mL×5);萃取液以氫氧化鈉溶液(1 mol/L)反萃取(30 mL×3),酸化反萃取液(pH 2~3),DCM萃取(35 mL×5),萃取液經飽和食鹽水洗滌,無水硫酸鈉干燥,過濾,濃縮得油狀物[18]。同法重復該反應3次;共得油狀物(5,1 140.6 mg,299.8 mCi,92%)。1H NMR(400 MHz,DMSO-d6):δ12.03(br,1H),7.63-6.98(m,5H),4.44(s,2H),3.49-3.39(m,2H),2.30-2.15(m,2H),1.66-1.42(m,4H)。ESI-MSm/z:211[M+H]+。
2.1.25-芐氧基[羰基-14C]戊酰胺(6) 在氬氣保護和0 ℃下,將5-芐氧基[羰基-14C]戊酸(5,227.4 mg)和N,N-二甲基甲酰胺(DMF,50 μL)溶于無水DCM(3 mL)中,攪拌;緩慢滴入草酰氯(418 mg)。滴畢攪拌10 min;升至室溫,攪拌1 h。減壓蒸除反應液中過量草酰氯,殘余物以無水DCM(3 mL)稀釋。在5~10 ℃下,將稀釋的酰氯溶液緩慢滴入氨飽和的干燥二氧六環(diox)溶液(2 mL)中,滴畢攪拌30 min;升至室溫,攪拌3.5 h。在線放射性高效液相色譜-二極管陣列檢測器/質譜聯用(HPLC-FSA/PDA/MS,簡稱HFPM)監測表明,原料(5)完全轉化。向反應液中加水(30 mL),減壓脫除有機溶劑,向殘余液中加入飽和碳酸氫鈉溶液(15 mL),以乙酸乙酯萃取(30 mL×5),萃取液經飽和食鹽水洗滌、無水硫酸鈉干燥、過濾、濃縮得淡黃色固體。同法重復該反應4次;共得淡黃色固體(6,943.6 mg,249.4 mCi,84%)。1H NMR(400 MHz,DMSO-d6):δ7.37-7.12(m,6H),6.65(s,1H),4.42(s,2H),3.48-3.33(m,2H),2.10-1.92(m,2H),1.64-1.29(m,4H)。ESI-MSm/z:210[M+H]+。
HPLC分析條件:SunFire C18色譜柱(5 μm,4.6 mm×250 mm);流速1.00 mL/min;波長 254 nm;柱溫 30 ℃;進樣量 5 μL;梯度洗脫(min/%A)控制:0/10,1/10,15/100,20/100,25/10,30/10,A為甲醇,B為0.05%甲酸水溶液。下文如無說明,HPLC分析均使用此條件。
2.1.35-羥基[羰基-14C]戊酰胺(7) 在氬氣保護下,將5-芐氧基[羰基-14C]戊酰胺(6,184.6 mg)、10%Pd/C(17 mg)和甲醇(4 mL)混合,反復抽真空-氫氣置換五次,向系統鼓入氫氣,在室溫常壓下攪拌。HFPM監測顯示,攪拌2 h后原料(6)反應完全。將反應混合物過濾,以甲醇淋洗濾餅5次,減壓濃縮濾液得白色固體[19]。同法重復該反應4次;共得白色固體(7,499.3 mg,230.7 mCi,94%)。1H NMR(400 MHz,DMSO-d6):δ7.24(br,1H),6.70(br,1H),3.43-3.32(m,1H),2.10-1.86(m,1H),1.64-1.28(m,1H)。ESI-MSm/z:120[M+H]+。
2.1.4[羰基-14C]戊二酰亞胺(8) 將硅膠(100~200目,720 mg)、氯鉻酸吡啶鎓鹽(PCC,680 mg)、5-羥基[羰基-14C]戊酰胺(7,125.7 mg)和無水THF(5 mL)的混合物超聲震蕩(內溫<40 ℃)[20]。HFPM監測表明,反應體系超聲2.5 h后(7)轉化完全。將反應混合物過濾,濾餅以THF淋洗3次,濃縮,濃縮物經快速硅膠層析(VMeOH∶VDCM=1∶100)得無色固體。同法重復該反應3次;共得白色固體(8,221.9 mg,106.3 mCi,47%)。1H NMR(400 MHz,DMSO-d6):δ10.64(s,1H),2.54-2.38(m,4H),1.89-1.77(m,2H)。ESI-MSm/z:116[M+H]+。
2.1.52,6-二氯[2-14C]吡啶(9) 在氬氣保護下,將[羰基-14C]戊二酰亞胺(8,107.6 mg)、五氯化磷(583 mg)和三氯化磷(2.5 mL)的混合液于室溫下攪拌。HFPM監測顯示,攪拌13 h后原料(8)完全轉化[21-22]。將反應液倒入劇烈攪拌的冰-水(50 mL)中,DCM萃取(40 mL×5),將萃取液依次用水、飽和碳酸氫鈉溶液和飽和食鹽水洗滌,無水硫酸鈉干燥,過濾后濃縮得白色固體。同法重復該反應1次;共得白色固體(9,248.3 mg,90.6 mCi,88%)。1H NMR(400 MHz,DMSO-d6):δ8.00-7.91(m,1H),7.64-7.54(m,2H)。ESI-MSm/z:150[M+H]+,152[M+2+H]+。
2.1.66-氯[2,6-14C]吡啶-2-醇(10) 在氬氣保護下,將2,6-二氯[2-14C]吡啶(9,99.2 mg)、氫氧化鉀(105 mg)和叔丁醇(2.5 mL)的混合液于90 ℃下攪拌。HFPM監測顯示,攪拌2.5 h后反應結束[23]。減壓脫除反應液中溶劑,加入水和石油醚(各50 mL),攪拌分層,以稀鹽酸酸化水層(pH 2~3),經乙酸乙酯萃取(40 mL×5)、飽和食鹽水洗滌和無水硫酸鈉干燥,過濾,濃縮得白色固體。同法重復該反應1次;共得白色固體(10,169.8 mg,71.5 mCi,83%)。1H NMR(400 MHz,DMSO-d6):δ11.44(br,1H),7.70-7.56(m,1H),6.95-6.81(m,1H),6.63-6.50(m,1H)。ESI-MSm/z:132[M+H]+,134[M+H]+。
2.1.73,5,6-三氯[2,6-14C]吡啶-2-醇(11) 在氬氣保護下,將次氯酸鈉溶液(1.35 mL,活性氯≥7.5%)加入攪拌的6-氯[2,6-14C]吡啶-2-醇(10,83.0 mg)和氫氧化鉀(40 mg)的水(1.50 mL)溶液中;在室溫下攪拌1.5 h反應結束[24]。將反應混合物降溫至0 ℃,以稀鹽酸酸化(pH 2~3),攪拌10 min,過濾,濾餅用純水洗滌3次,在40 ℃真空干燥后得淡黃色固體。同法重復該反應1次;共得淡黃色固體(11,149.0 mg,40.8 mCi,58%)。1H NMR(400 MHz,DMSO-d6):δ12.96(br s,1H),8.26(s,1H)。ESI-MSm/z:200[M+H]+,202[M+2+H]+,204[M+4+H]+。
2.1.8目標物14C-毒死蜱(1,O,O-二乙基-O-(3,5,6-三氯-2-[2,6-14C]吡啶基)硫代磷酸酯) 在氬氣保護下,將二乙基硫代磷酰氯(165 mg)加入3,5,6-三氯[2,6-14C]吡啶-2-醇(11,146.3 mg)和碳酸鈉(93 mg)的干燥DMF(3 mL)中,室溫攪拌。HFPM監測顯示,攪拌2 h原料(11)消耗完全[14,25]。以稀鹽酸酸化反應液(pH 2~3),加水(30 mL),以乙酸乙酯-石油醚混合液(1∶1,V/V)萃取(35 mL×5),萃取液經飽和食鹽水洗滌(40 mL),無水硫酸鈉干燥,過濾后濃縮得粗品。粗品經制備型RP-HPLC純化得白色固體(1,193.1 mg,30.2 mCi,75%)。1H NMR(400 MHz,DMSO-d6):δ8.62(s,1H),4.42-4.14(m,4H),1.32-1.25(m,6H)。ESI-MSm/z:352[M+H]+,354[M+2+H]+,356[M+4+H]+。制備色譜條件:xBridge Prep C18柱(10 μm,150 mm×19 mm),流速7.00 mL/min,波長270 nm,進樣量500 μL;柱溫30 ℃;梯度洗脫(min/%A)控制:0/30,1/30,18/40,21/0,24/0,27/30,30/30;A為水,B為甲醇。收集保留時間為15.450~16.423 min的組分。
2.2 同位素標記化合物的質量指標分析
2.2.1放化純度的測定 白色固體(1)的放化純度分別采用放射性薄層成像分析(TLC-IIA)、離線放射性高效液相色譜(HPLC-LSC)和在線放射性高效液相色譜(HPLC-FSA)法測定[26-27]。
(1) TLC-IIA分析:將白色固體(1)和毒死蜱標樣在同一硅膠薄板(20 cm×5 cm)上連續展開4次;TLC條件:二氯甲烷/石油醚(VMeOH∶VDCM=1∶100)。在UV燈下,在該層析板上兩個樣品均顯示一個規則的斑點(Rf=0.64);將該層析板進行成像,該硅膠板上僅顯示一個規則的放射性斑點(Rf=0.64)。
(2) HPLC-FSA分析:準確稱取9.63 mg白色固體(1),以甲醇溶解并定容至100.00 mL,得母液。將母液稀釋至10 μg/mL,取10 μL進行HPLC-FSA分析。流動液閃采用Optiphase HiSafe 3閃爍液,流速為8 mL/min。白色固體(1)在HPLC-FSA色譜圖中均顯示1個色譜峰,保留時間為7.318 min。HPLC色譜條件:SunFire C18色譜柱(5 μm,4.6 mm×150 mm,美國沃特世公司);梯度洗脫(min/%A)控制:0/45,1/45,15/100;其余條件同2.1.2節。
(3) HPLC-LSC分析:取10 μL白色固體(1)稀釋液(10 μg/mL)進行HPLC分析,用自動接收器收集洗脫液,每隔20 s收集1份,用LSC分別測定各收集液活度。HPLC色譜條件同HPLC-FSA分析。根據測定結果繪制(1)的離線放射性高效液相色譜圖,結果如圖3所示。在圖3中,保留時間為420~460 s之間呈現1個放射性色譜峰(對應收集液為第21~23份)。根據該色譜峰對應物質的總活度與10 μL白色固體(1)稀釋液(10 μg/mL)活度的比值即放化純度;平行分析3次,取其平均值作為白色固體(1)的放化純度。
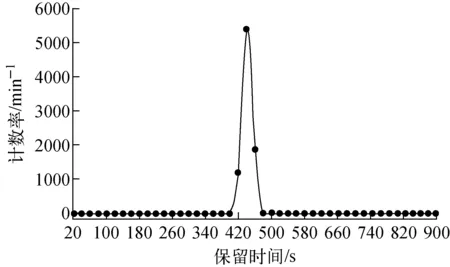
圖3 放射性同位素碳-14標記毒死蜱(1)的離線放射性高效液相色譜圖(HPLC-LSC)Fig.3 Offline radio-chromatogram (HPLC-LSC) of the carbon-14 labelled chlorpyrifos (1)
2.2.2化學純度的測定 白色固體(1)屬于高比活度標記物,采用HPLC-UV色譜圖中色譜峰面積歸一法確定其化學純度。結果如圖4所示,(1)的保留時間為7.351 min。色譜條件同2.2.1節。
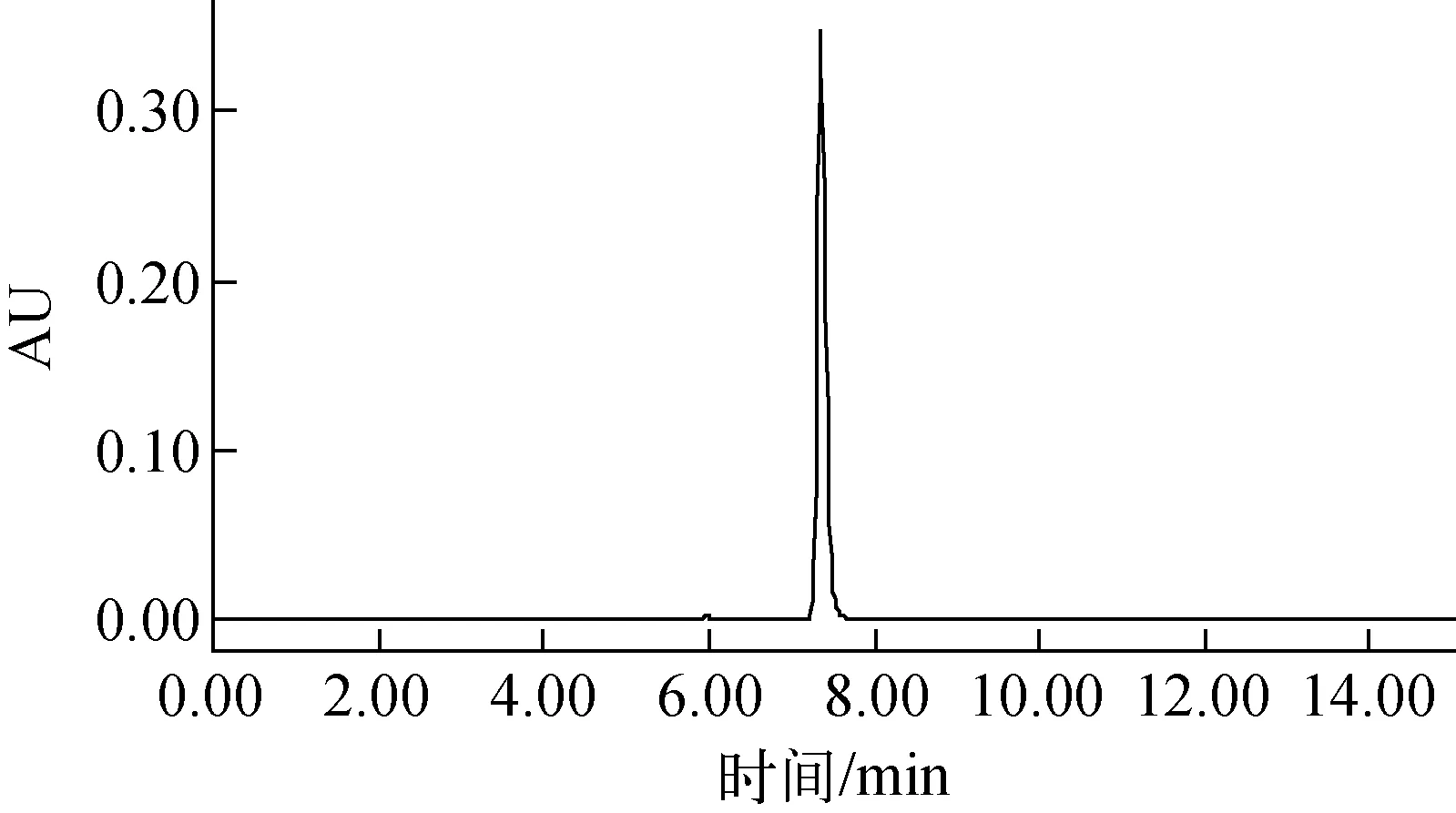
圖4 放射性同位素碳-14標記毒死蜱(1)的高效液相色譜圖(254 nm)Fig.4 High performance liquid chromatogram of radioisotope carbon-14 labelled chlorpyrifos (254 nm)
2.2.3比活度的測定 白色固體(1)的比活度按照文獻方法測定[26]。
3 結果與討論
以[14C]碳酸鋇為放射性同位素原料,通過格氏反應、親核取代、分子內環化反應、脫水芳構化、親電氯代等9步放化反應獲得碳-14標記毒死蜱粗品,經反相高效液相色譜純化獲得白色固體(1,30.2 mCi,總放化收率為11%)。白色固體(1)的核磁共振氫譜與毒死蜱標樣一致;HPLC-FSA/PDA/MS分析顯示(1)的相對分子質量比毒死蜱大2,且放射性高效液相色譜保留時間與毒死蜱標樣的HPLC保留時間一致。據以上分析結果并結合標記物的合成技術路線可確定,白色固體(1)為目標物碳-14標記毒死蜱(O,O-二乙基-O-(3,5,6-三氯-2-[2,6-14C]吡啶基)硫代磷酸酯)。TLC-IIA、HPLC-LSC、HFPM和LSC分析表明,標記物(1)的放化純度和化學純度均大于98%,比活度為55.2 mCi/mmol,該質量指標滿足下游示蹤試驗的要求,因而標記物(1)可作為放射性示蹤劑,可替代文獻中報道的標記物(O,O-二乙基-O-(3,5,6-三氯-2-[2,6-14C2]吡啶基)硫代磷酸酯[14]),用于毒死蜱的藥代動力學、生態環境安全與污染消減機制等問題的研究。
碳-14是利用同位素示蹤法研究農醫藥的代謝與安評中常用的核素,合成碳-14標記農醫藥首先需要考慮的是標記位置的選擇[12-13]。需要指出的是,選用不同的標記位置的碳-14標記物,則下游示蹤試驗的結果往往有所不同;而碳-14標記位置選擇不當,則會引起下游試驗結果與客觀情況有所偏差,甚至導致結論謬誤。例如,文獻報道的乙基碳-14標記毒死蜱(2,O,O-二[1-14C]乙基-O-(3,5,6-三氯-2-吡啶基)硫代磷酸酯,見圖1)合成步驟少,成本低,但其中[1-14C]乙氧基標記片段通過磷酸酯鍵與吡啶環相連,因其易從分子骨架脫落而無法滿足生物體(植物、哺乳動物和家禽)、水體-土壤環境中大多數代謝試驗對示蹤劑的要求[28]。
在用于代謝試驗的碳-14標記農醫藥合成中,通常根據農醫藥母體分子的化學結構將其劃分為若干標記單元(即分子中標記相對牢固的位置),然后依據標記合成的必要性、可行性、標記難度及實驗目的和要求在各個標記單元內選擇恰當的位點進行標記。對于含一個標記單元的農藥,僅需對標記單元中任意一個位點的碳原子進行標記;對于含兩個或多個標記單元的農藥,則需要對各個標記單元分別進行標記,標記位點可選擇標記單元中任意一個碳原子[6]。考慮到研究成本和實驗目的,筆者均從每種農藥中均選取兩個標記單元分別進行標記,且以芳環標記為主[6-10,17,26-27];而僅有少數碳-14標記物,如毒死蜱、精草銨膦、利谷隆、氟啶蟲酰胺、丁噻隆和特丁津,均含一個標記單元,僅需選取標記單元內任意一個位點進行標記。碳-14是自旋量子數為0的非磁性核,無核磁共振現象,因而無法利用核磁共振碳譜直接確定碳-14標記物中碳-14的位點。盡管綜合利用核磁共振定量碳譜、質譜和液體閃爍測量等技術可間接地推測碳-14的位點,但因分析過程所需樣品較大而放射性標記物通常均為微量,因而確定未知來源的放射性物質中碳-14位點往往難以實現[29]。當前,國際上確定碳-14標記物中碳-14位點的最可靠方法是將標記合成技術路線與其他分析手段相結合,綜合分析確定。
香草硫縮病醚(12)、毒死蜱(13)和氟啶蟲酰胺(14)分子中的標記單元示于圖5。本研究選取具有芳香性的吡啶環為標記位置,以吡啶環中2,6-位碳為標記位點,這種標記較為牢固,碳-14不易從吡啶環脫落,可滿足大多數示蹤試驗的要求。相比較而言,采用標記生物大分子常用的核素碘-131標記毒死蜱,合成簡單,成本低,但碳-14標記毒死蜱(1)的理化性質和生物學性質更貼近毒死蜱,標記物的物理半衰期更長,穩定性更高,因而更適合于下游示蹤試驗[30]。

圖5 香草硫縮病醚(12)、毒死蜱(13)和氟啶蟲酰胺(14)分子中的標記單元Fig.5 Radiolabelling units of XCLSBM (12), chlorpyrifos (13) and flonicamid (14)
文獻報道碳-14標記毒死蜱(O,O-二乙基-O-(3,5,6-三氯-2-[2,6-14C2]吡啶基)硫代磷酸酯)制備方法中起始同位素原料[14C]氰化鉀為劇毒品,且[14C]氰化鉀與1,3-二溴丙烷制備[1,4-14C2]戊二腈的親核取代反應非常緩慢,長達65 h;[1,4-14C2]戊二腈在210 ℃高溫下反應18 h方可制得[1,4-14C2]戊二酰亞胺,因而反應條件非常苛刻[14]。在本研究中,以[14C]二氧化碳在低溫下進行反應,經過3步接近常溫常壓條件制備了[1-14C]戊二酰亞胺,反應速度較快,且反應易于控制。文獻報道中間體[2,6-14C2]氯吡啶必須用劇毒的氯氣在高壓釜內高溫進行氯化,操作具有潛在的危險性,而本研究在室溫常壓條件下采用次氯酸體系對[2,6-14C]三氯吡啶醇(10)進行氯化,進而通過在堿性條件下與二乙基硫代磷酰氯反應而獲得目標物(1)。
4 結論
以[14C]碳酸鋇為放射性同位素原料,通過9步放化反應獲得碳-14標記毒死蜱(1,O,O-二乙基-O-(3,5,6-三氯-2-[2,6-14C]吡啶基)硫代磷酸酯,30.2 mCi),反應總收率為11%。該標記物的技術指標滿足下游同位素示蹤試驗的要求,因而它可作為放射性示蹤劑,用于毒死蜱的藥代動力學、生態環境安全與污染消減機制等問題的研究。
