稀釋劑分子結(jié)構(gòu)對TBP-Zr(NO3)4-HNO3萃取體系極限有機(jī)相濃度的影響
2020-12-21 10:26:42王文濤張春龍謝書寶袁潔瓊
濕法冶金 2020年6期
關(guān)鍵詞:體系
王文濤,曹 智,張春龍,謝書寶,袁潔瓊,蘇 哲
(1.中國原子能科學(xué)研究院 放射化學(xué)研究所,北京 102413;2.生態(tài)環(huán)境部 核與輻射安全中心,北京 102488)
在乏燃料后處理領(lǐng)域,普雷克斯(PUREX)流程是當(dāng)前商業(yè)化運(yùn)行技術(shù)之一。該技術(shù)以磷酸三丁酯(TBP)作萃取劑,通過多級逆流萃取使U、Pu與裂變產(chǎn)物分離[1]。TBP萃取劑具有選擇性高、對裂變產(chǎn)物凈化系數(shù)高等優(yōu)點(diǎn),也具有化學(xué)/輻照穩(wěn)定性好、沸/閃點(diǎn)高、揮發(fā)性低、水中溶解度低等優(yōu)勢;但TBP密度與水密度接近,黏度較大,萃取過程中會(huì)使有機(jī)相與水相難以分開;因此,需加入稀釋劑來降低其密度和黏度,改善水力學(xué)性能[2-3]。稀釋劑加入后,有機(jī)相中的萃合物濃度超過一定量時(shí),有機(jī)相會(huì)發(fā)生相分離,形成上層富含稀釋劑的輕相和下層富含萃合物的重相,其中下層重相通常被稱作“第三相”[4]。第三相的出現(xiàn)不利于萃取工藝的連續(xù)穩(wěn)定運(yùn)行,特別是在快堆乏燃料后處理時(shí),Pu(Ⅳ)在第三相的富集可能會(huì)有核臨界風(fēng)險(xiǎn),因此必須設(shè)法避免[5]。
通常,把剛好出現(xiàn)第三相時(shí)有機(jī)相中金屬萃合物濃度稱為極限有機(jī)相濃度(limiting organic concentration,LOC)。LOC是衡量稀釋劑性能的關(guān)鍵參數(shù)之一,對于給定的萃取體系,其大小主要由稀釋劑的性質(zhì)決定[6]。
國內(nèi)外學(xué)者針對稀釋劑的研究發(fā)現(xiàn),平均碳原子數(shù)為12的惰性烷烴(如正十二烷、煤油或其他C10~C14的烷烴混合物)稀釋劑可滿足Purex流程要求[7-8]。烷烴稀釋劑的LOC與其分子結(jié)構(gòu)密切相關(guān):對于正構(gòu)烷烴,碳數(shù)越大,LOC越小;對于同碳數(shù)烷烴,異構(gòu)化有助于提高LOC,異構(gòu)化程度越高,其LOC越高[9-10]。法國的阿格和馬庫爾后處理廠采用高度支鏈化的氫化四聚丙烯(TPH,平均碳數(shù)為12)作稀釋劑,印度的特朗貝后處理廠采用Shellsol-T異構(gòu)烷烴作稀釋劑,都有很好效果,對U、Pu關(guān)鍵核素的負(fù)載能力和輻照穩(wěn)定性都顯著優(yōu)于傳統(tǒng)的正十二烷和煤油。所以,異構(gòu)烷烴也被認(rèn)為是Purex流程的優(yōu)選稀釋劑[11]。然而,由于TPH是由近百種異構(gòu)烷烴組成的復(fù)雜混合物,所以其具體作用機(jī)制仍不清楚。在Th(NO3)4-TBP-HNO3體系中,稀釋劑碳鏈越短、支化度越高,其LOC越大[10];在C923-Ir(IV)-HCl體系中,稀釋劑不同,LOC由大到小順序?yàn)榧妆健侄妆?環(huán)己烷>正辛烷>正壬烷>煤油>正十二烷[12];在TBP萃取無機(jī)酸時(shí)也有類似稀釋劑效應(yīng)[13]。但總體而言,針對稀釋劑的研究仍主要集中在直鏈烷烴和少數(shù)幾種商業(yè)化異構(gòu)烷烴,有關(guān)烷烴稀釋劑取代基種類、數(shù)量和位置等結(jié)構(gòu)信息與LOC之間的關(guān)系仍很模糊,系統(tǒng)研究十分必要。
試驗(yàn)針對TBP-Zr(NO3)4-HNO3體系,研究了稀釋劑烷烴碳數(shù),取代基種類、數(shù)量及位置等結(jié)構(gòu)參數(shù)對LOC的影響,并在此基礎(chǔ)上考察不同組成混合烷烴稀釋劑的LOC的差異,以期為液-液萃取體系稀釋劑的選用與制備提供技術(shù)支持。
1 試驗(yàn)部分
1.1 主要試劑
正構(gòu)烷烴稀釋劑購自試劑公司,其余稀釋劑自行設(shè)計(jì)、委托合成。稀釋劑的分子結(jié)構(gòu)、來源和規(guī)格見表1。
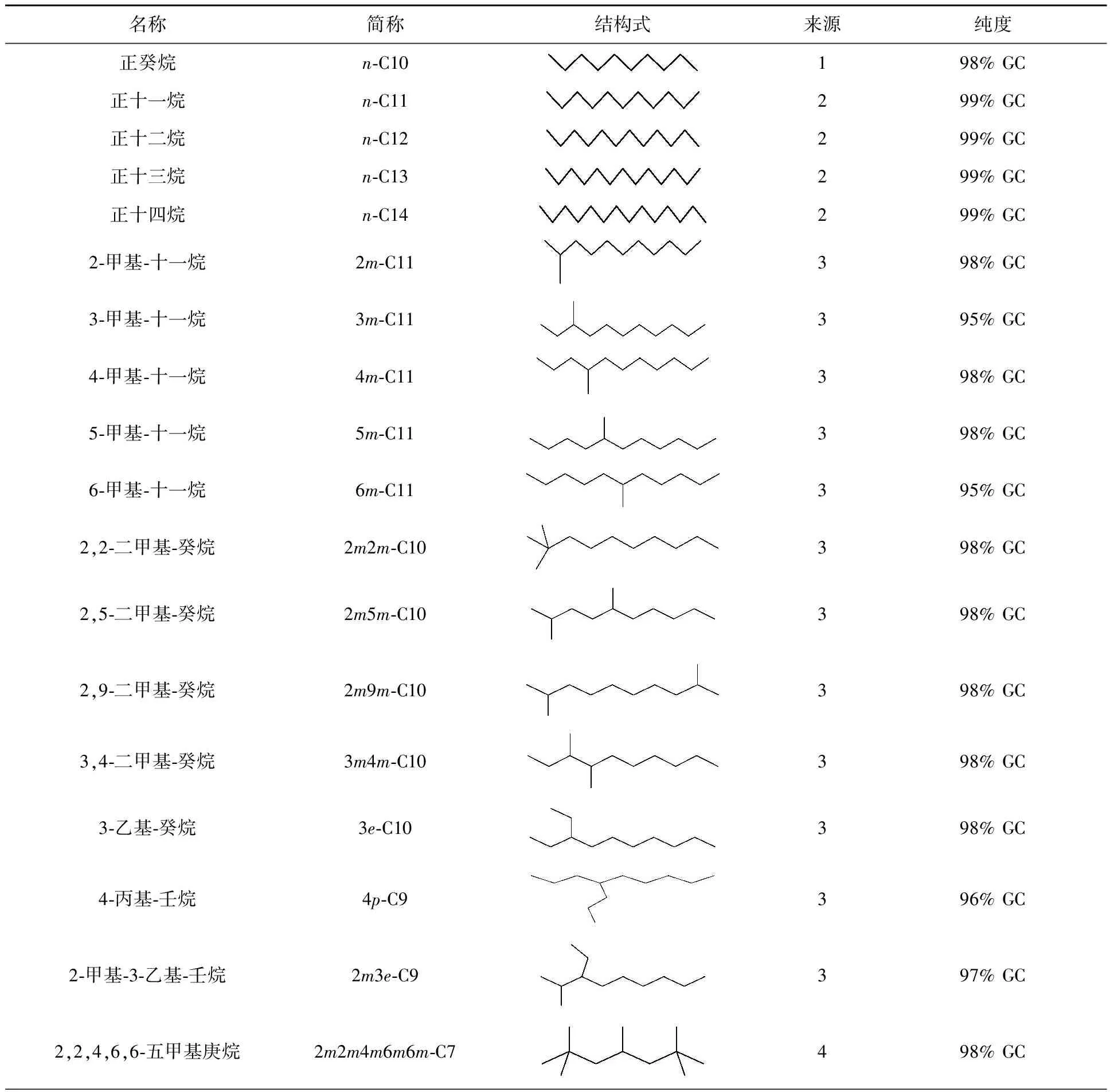
表1 稀釋劑的來源及規(guī)格
磷酸三丁酯(TBP),純度99%,北京百靈威科技有限公司;Zr(NO3)4,分析純,北京化學(xué)試劑公司;濃HNO3(65%~68%),國藥集團(tuán)化學(xué)試劑公司,使用前用Mill-Q 18.2 MΩ超純水稀釋,濃度用NaOH標(biāo)準(zhǔn)溶液滴定測定。
1.2 稀釋劑的組成
加氫煤油、Mineral Spirits、稀釋劑1~3等混合烷烴組成及分布采用氣相色譜-質(zhì)譜聯(lián)用儀(GC-MS)進(jìn)行測定。
儀器及操作條件:Agilent 7890A/5977MSD,色譜柱(DB-5MS,30 m×0.25 mm×0.25 μm);初始溫度60 ℃,保持2 min后以10 ℃/min速度升至250 ℃;進(jìn)樣口溫度250 ℃,傳輸線溫度250 ℃;流量1 mL/min,進(jìn)樣量0.2 μL,分流比200/1;離子源溫度220 ℃;EI電離源,能量70 eV。
異構(gòu)烷烴的組成:采用保留指數(shù)(RI)作為區(qū)分烷烴樣品的碳數(shù)及單、雙、多取代組分的依據(jù)[14-15],并利用isoparaffin軟件(中石化石油化工科學(xué)研究院開發(fā),版本1.0)統(tǒng)計(jì)分析各組分碳數(shù)及取代基情況。
1.3 LOC的測定
將不同稀釋劑與TBP配制成30%混合溶劑,依次用等體積5%碳酸鈉和0.1 mol/L HNO3溶液洗滌2次,然后用等體積6 mol/L HNO3溶液平衡3次。
LOC測定采用加貧有機(jī)相法。首先,在離心管中加入1 mL酸平衡3次的混合溶劑,然后加入1 mL質(zhì)量濃度為23.47 g/L的濃硝酸鋯溶液(硝酸濃度6 mol/L),劇烈振蕩(轉(zhuǎn)速2 500 r/min)3 min,之后在離心機(jī)(湘儀TD-4型,轉(zhuǎn)速3 000 r/min)中離心分相3 min,此時(shí)有機(jī)相分相出現(xiàn)第三相;隨后,在有機(jī)相中加入少量酸平衡3次后的混合溶劑,振蕩、離心、觀察第三相是否消失(通常需要反復(fù)多次上述操作才會(huì)使第三相消失);最后,分別取第三相剛好消失時(shí)的有機(jī)相和水相,稀釋后進(jìn)行ICP-AES測定。所有試驗(yàn)均在(20±1)℃下進(jìn)行。
2 試驗(yàn)結(jié)果與討論
萃取劑分子通常具有一定的兩親性。一般認(rèn)為,萃合物在稀釋劑中不是以真溶液形式存在,而是以反膠束或微乳液等聚集形式存在[16]。反膠束是液-液萃取中一種被普遍接受的模型,該模型認(rèn)為反膠束的極性核是由萃取劑的極性頭基及金屬、酸或水等極性物質(zhì)組成,而其疏水外層則主要由萃取劑的烷基鏈組成。反膠束的極性核之間存在較強(qiáng)的范德華力(吸引),疏水外殼中烷基鏈之間則存在位阻穩(wěn)定作用(排斥),從而使反膠束之間實(shí)現(xiàn)微觀力學(xué)平衡。熱力學(xué)上,內(nèi)核親水、外層疏水的反膠束結(jié)構(gòu)能有效降低體系自由能,使體系分散得很均勻;但當(dāng)金屬、酸等極性物質(zhì)更多地進(jìn)入反膠束極性內(nèi)核時(shí),極性核之間的范德華力也相應(yīng)增大。研究表明,當(dāng)極性核之間的吸引力超過反膠束平均熱運(yùn)動(dòng)能kBT(kB為Boltzmann常數(shù))的2倍時(shí),反膠束之間會(huì)傾向于融合而發(fā)生相分離,宏觀上即形成第三相[6,17]。
稀釋劑和萃取劑分子結(jié)構(gòu)對反膠束之間的相互作用有重要意義,稀釋劑與萃取劑之間的分子間相互作用越強(qiáng),越不容易發(fā)生相分離,即LOC越大。稀釋劑分子鏈越短,就越易滲透到反膠束非極性區(qū)域,降低膠束極性核之間的范德華吸引力,從而起到溶脹和穩(wěn)定反膠束的作用[18];稀釋劑分子鏈越長,這種作用就越不明顯。而當(dāng)稀釋劑或萃取劑分子鏈出現(xiàn)支化結(jié)構(gòu)時(shí),分子間的位阻穩(wěn)定作用增強(qiáng)有利于反膠束的穩(wěn)定,這就是異構(gòu)烷烴的LOC比同碳數(shù)正構(gòu)烷烴大的原因。因此,也可利用反膠束相互作用模型來解釋不同結(jié)構(gòu)烷烴稀釋劑在TBP-Zr(NO3)4-HNO3萃取體系中LOC的差異。
2.1 烷烴主鏈碳數(shù)對LOC的影響
TBP-Zr(NO3)4-HNO3萃取體系中,以不同結(jié)構(gòu)烷烴為稀釋劑時(shí)的LOC見表2,烷烴主鏈碳原子數(shù)與LOC之間的關(guān)系如圖1所示。
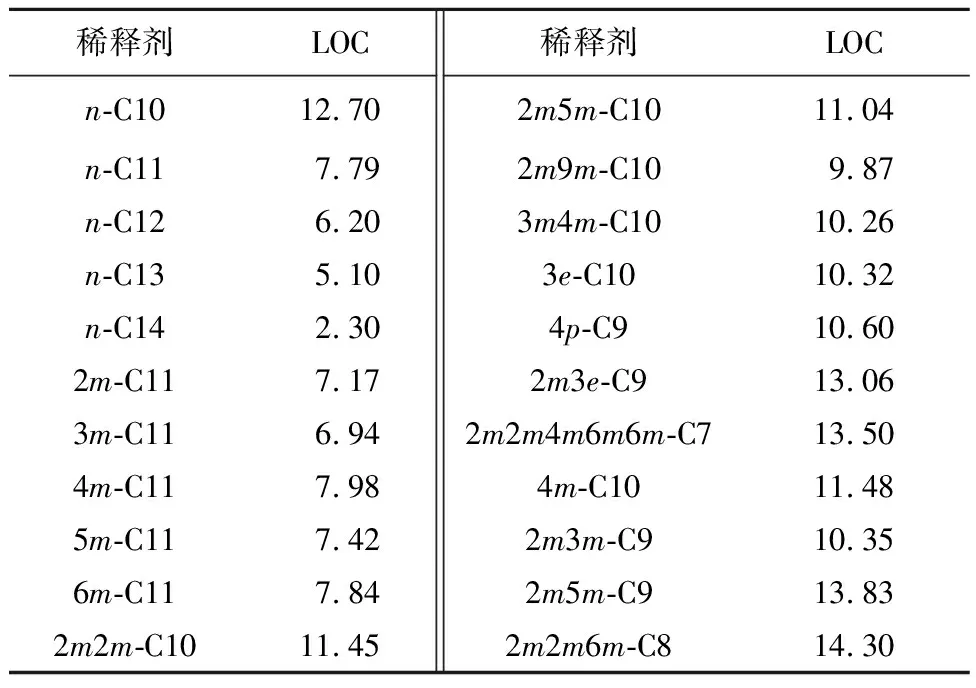
表2 TBP-Zr(NO3)4-HNO3萃取體系中不同稀釋劑對應(yīng)的LOC
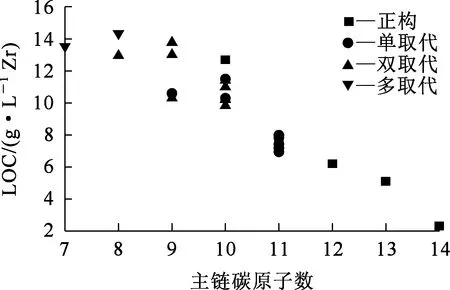
圖1 烷烴主鏈碳原子數(shù)與LOC之間的關(guān)系
由圖1看出:對于正構(gòu)烷烴來說,其LOC隨碳數(shù)增加而降低。這可能是由短鏈稀釋劑分子更易穿透到反膠束的非極性外層,進(jìn)而穩(wěn)定反膠束引起。類似的規(guī)律也在TBP-U(Ⅵ)/Th(Ⅳ)/Pa(Ⅴ)等萃取體系中存在[13,18-19]。對于異構(gòu)烷烴,情況更復(fù)雜:5種單甲基取代的C11烷烴(2mC11、3mC11、4mC11、5mC11、6mC11)的LOC均小于所有C10及以下烷烴的LOC;但C7~C9取代烷烴的LOC與主鏈碳數(shù)間的關(guān)系較復(fù)雜,除2個(gè)C9烷烴(4p-C9和2m3m-C9)低于C10的LOC,及2m2m4m6m6m-C7的LOC比2m2m6m-C8的略低之外,其余烷烴都能符合隨主鏈碳數(shù)增加而LOC降低的規(guī)律。因此,可以認(rèn)為,主鏈碳數(shù)對烷烴稀釋劑的LOC有較大影響,無論是正構(gòu)烷烴還是異構(gòu)烷烴,可近似認(rèn)為其LOC與主鏈碳數(shù)成反比。
2.2 取代基性質(zhì)(種類、數(shù)量和位置)對LOC的影響
由表2看出:當(dāng)烷烴總碳數(shù)相同時(shí),取代基的種類、數(shù)量和位置不同會(huì)使其LOC發(fā)生較為顯著變化。n-C10、n-C11和C12烷烴與LOC之間的關(guān)系如圖2所示。
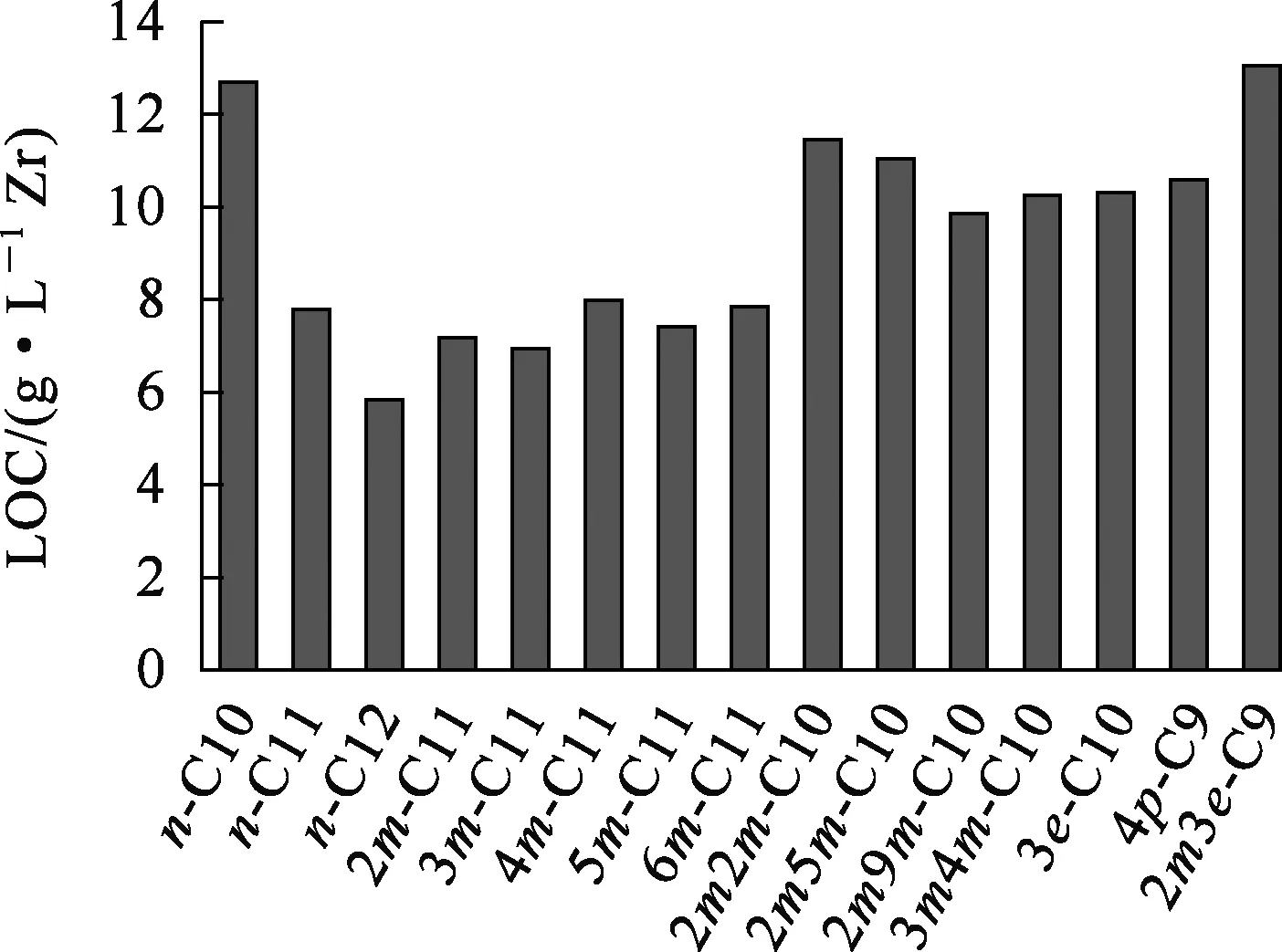
圖2 n-C10、n-C11和C12烷烴與LOC之間的關(guān)系
由圖2看出:取代烷烴的LOC均大于同碳數(shù)正構(gòu)烷烴的LOC;隨取代基數(shù)量增加,其LOC也增大。其中5種單甲基取代的C11的LOC接近或略小于n-C11的LOC,且甲基位置對其LOC有輕微影響;對于2m-C11來講,導(dǎo)致這一現(xiàn)象的原因可能是2位C原子被甲基取代后,烷烴分子的一端體積增大,不利于其與反膠束中TBP的烷基鏈相互作用;3位C原子被甲基取代后,其端基體積更大,所以3m-C11的LOC進(jìn)一步降低;而當(dāng)4位以后C原子被甲基取代后,這種體積效應(yīng)則不明顯。
類似的體積效應(yīng)在雙甲基取代的C10烷烴中也存在:2m9m-C10的LOC明顯低于其他雙甲基,這可能是2m9m-C10分子中2個(gè)甲基分別在烷烴鏈兩端的2位C原子上,這使2m9m-C10分子兩頭體積較大,空間位阻上不利于進(jìn)入反膠束的外層的原因。
取代基數(shù)量繼續(xù)增加,即2m2m4m6m6m-C7和2m2m6m-C8,其LOC相對于n-C12大幅增大,這可能是碳鏈大幅縮短的緣故。所以,實(shí)際工藝中,選用多取代含量高的稀釋劑效果會(huì)更好。
取代基的種類也有明顯影響:2m3e-C9的LOC比2m3m-C9大(見表2),顯然這是由3位碳原子上取代基長度不同引起的。其原因可能是乙基能部分參與穩(wěn)定反膠束,但準(zhǔn)確機(jī)制仍需深入研究。
由上可知,為提高萃取分離效率(高LOC),應(yīng)盡量選擇碳原子數(shù)少的直鏈烷烴作稀釋劑;但碳原子數(shù)少的直鏈烷烴閃點(diǎn)低,在使用作用中容易引發(fā)安全事故,所以必須確保稀釋劑分子足夠大(碳原子數(shù)足夠多),以確保其安全性。當(dāng)主鏈碳原子數(shù)確定時(shí),盡量選擇二取代及多取代烷烴占比高的稀釋劑。
2.3 不同來源混合烷烴稀釋劑的LOC
實(shí)際生產(chǎn)中,出于成本考慮,后處理廠不可能采用純的異構(gòu)烷烴作為稀釋劑,已經(jīng)應(yīng)用的異構(gòu)烷烴稀釋劑也均是混合物[20]。因此,有必要研究稀釋劑組成對LOC的影響規(guī)律。試驗(yàn)選取工業(yè)上常用的加氫煤油OK、商業(yè)化異構(gòu)烷烴混合物MS,及定制的3種不同組成的烷烴混合物。5種稀釋劑的GC-MS圖譜如圖3所示。不同烷烴的保留指數(shù)不同,根據(jù)保留指數(shù)并結(jié)合質(zhì)譜數(shù)據(jù)得到這5種稀釋劑的碳數(shù)分布和各類異構(gòu)烷烴占比,結(jié)果見表3。
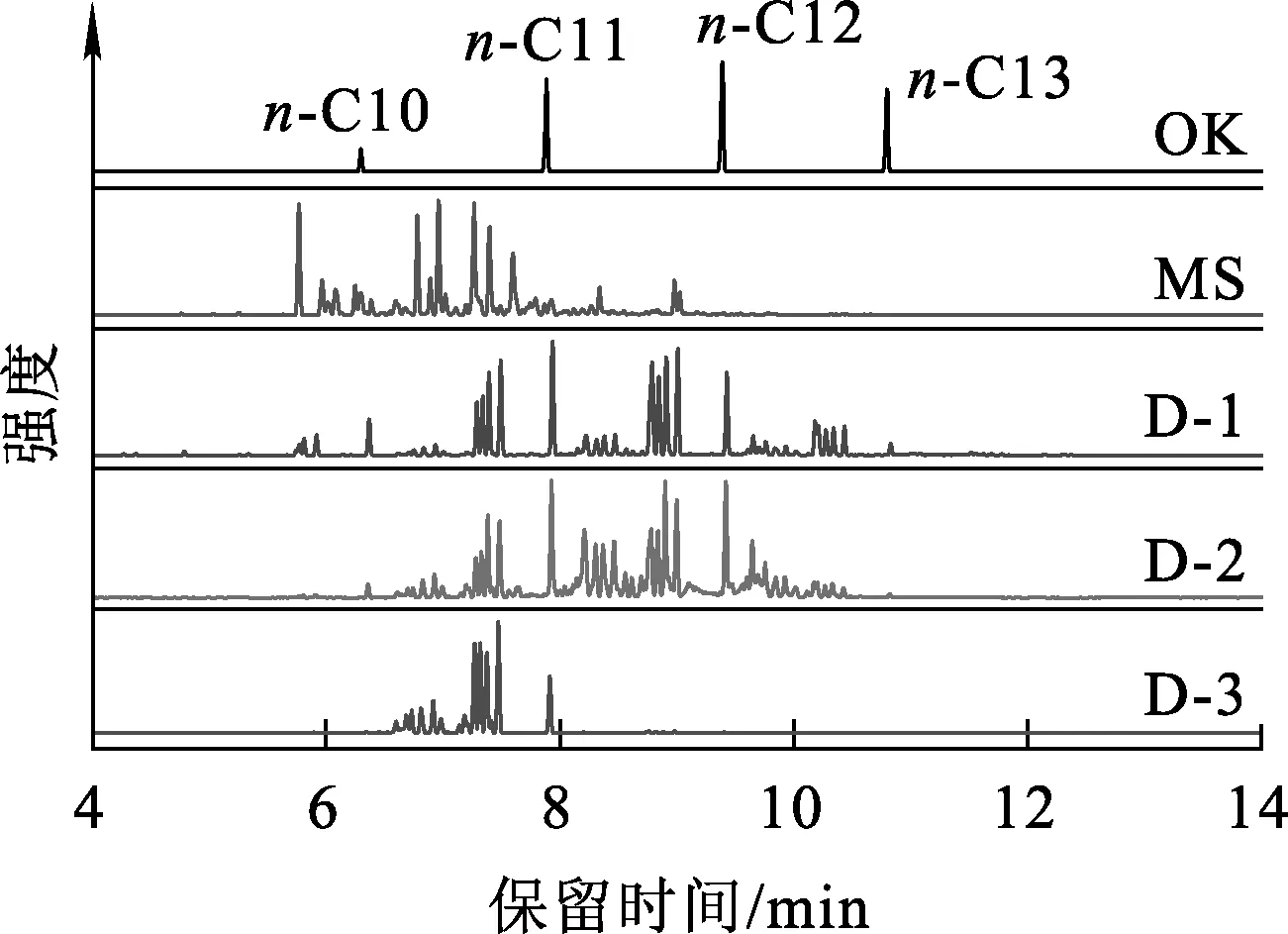
圖3 5種混合烷烴稀釋劑的GC-MS圖譜
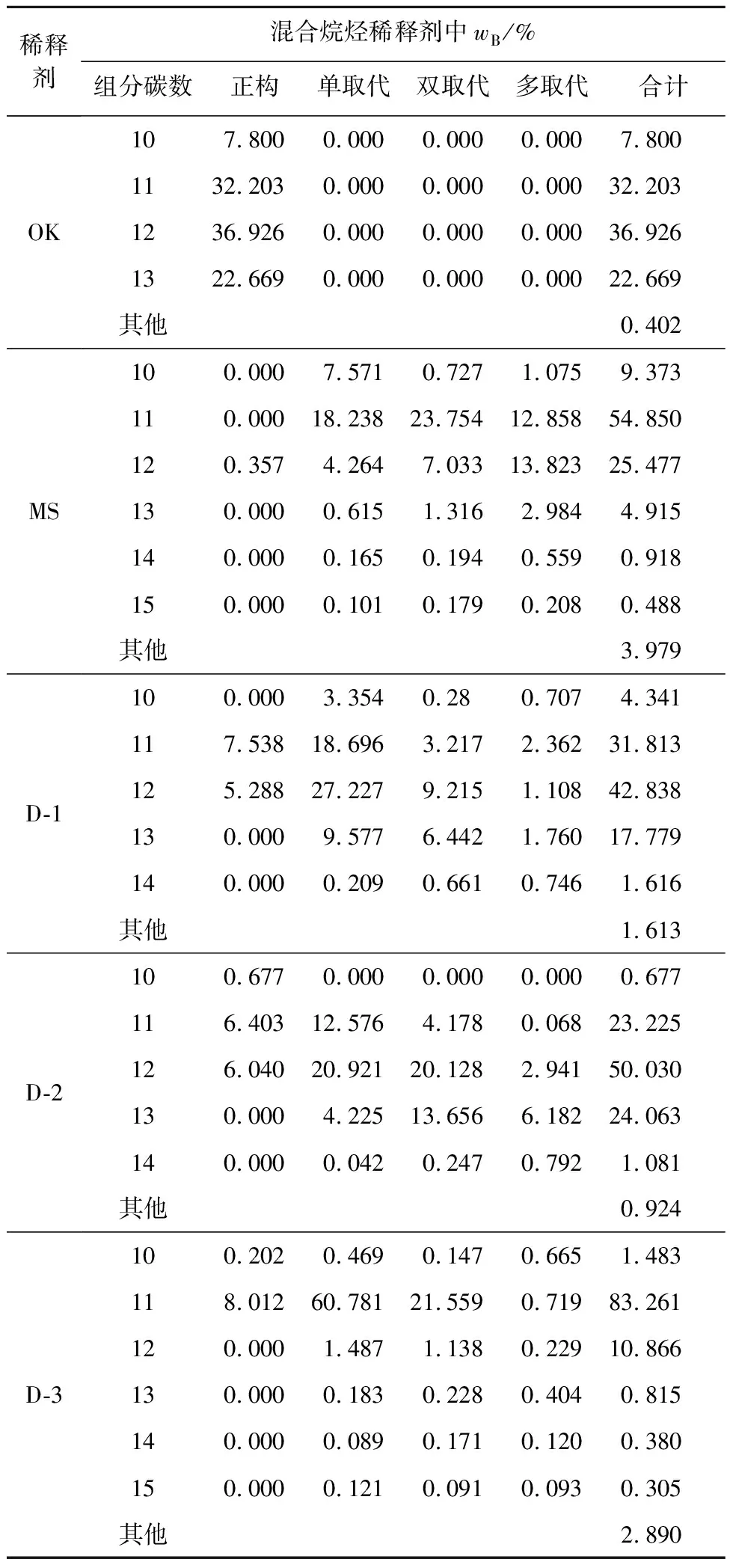
表3 不同混合烷烴稀釋劑的組成及分布
由表3看出:OK由4個(gè)組分組成,可依次歸屬于C10~C13等正構(gòu)烷烴;MS譜圖較為復(fù)雜,其出峰時(shí)間較為靠前,其中C11組分占比超過50%,且各碳數(shù)組分中雙取代和多取代占比高;D-1和D-2均由10%左右的正構(gòu)烷烴和90%左右的異構(gòu)烷烴組成,但其組成和分布有較大差異,其中D-1的低碳組分占比較D-2的高,但D-2中C12以上的雙取代和多取代組分占比又較D-1的高;D-3主要是由C11組分構(gòu)成,且以單、雙取代為主。
5種混合稀釋劑組成的萃取體系的LOC如圖4所示。結(jié)合表3看出:稀釋劑中正構(gòu)烷烴占比越小,LOC越大;稀釋劑中低碳組分占比越大,LOC越大;雙取代和多取代烷烴占比越大,LOC越大。

圖4 5種混合烷烴稀釋劑組成的萃取體系的LOC
3 結(jié)論
以TBP-Zr(NO3)4-HNO3萃取體系為模型體系,研究系列不同主鏈碳原子數(shù)、取代基種類、數(shù)量和位置的烷烴稀釋劑,得到以下結(jié)論:
1)烷烴主鏈碳原子數(shù)是影響其LOC的首要因素,主鏈碳原子數(shù)越小,LOC越大;
2)烷烴取代基的性質(zhì)(種類、數(shù)量和位置)對LOC有較大影響。烷烴碳原子數(shù)一定時(shí),取代基越多,LOC越大;取代基位置靠近分子鏈內(nèi)部且分布在一端時(shí)有利用提高LOC。
3)利用反膠束模型可以較好解釋稀釋劑的結(jié)構(gòu)效應(yīng),短鏈稀釋劑能較好進(jìn)入TBP反膠束疏水外殼,起到溶脹和穩(wěn)定反膠束作用,而長鏈稀釋劑則較難進(jìn)入反膠束外殼,這種穩(wěn)定化作用隨鏈長增加而不明顯;當(dāng)烷烴上出現(xiàn)取代基時(shí),凡是有利于稀釋劑分子和TBP的烷烴鏈相互作用的因素,均有利于穩(wěn)定反膠束,宏觀上表現(xiàn)出較高的LOC。
4)在滿足閃點(diǎn)等理化指標(biāo)要求前提下,選用低碳組分多及雙取代和多取代含量高的烷烴有利于提高稀釋劑的負(fù)載能力。
猜你喜歡
商品與質(zhì)量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀(jì)智能(數(shù)學(xué)備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛(wèi)生(2015年12期)2015-11-10 05:13:40
現(xiàn)代企業(yè)(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
