五種治療阿爾茨海默病藥物分子結構與性質的密度泛函計算分析
2020-12-28 13:11:36嚴茜茜黃羅儀王朝杰向錚
溫州醫科大學學報 2020年12期
嚴茜茜,黃羅儀,王朝杰,向錚
(1.溫州醫科大學 藥學院,浙江 溫州 325035;2.溫州市第七人民醫院 藥劑科,浙江 溫州 325000)
阿爾茨海默病(Alzheimer’s disease,AD)是一種起病潛隱,病癥呈進程性漸進的神經退行性疾病,具有記憶衰退,認知惡化,執行能力喪失以及行為改變等特性。在高齡人群中易生成癡呆,占全部癡呆類型的近60%[1],嚴重降低當代老年人生活質量。AD病理復雜并伴多種代謝途徑共同參與[2],其發病機制假說眾多,主要有膽堿能神經元損傷機制[3],β-淀粉樣蛋白級聯機制[4]、τ蛋白過度磷酸化機制。2020年4月,美國南加州大學凱克醫學院的研究團隊又提出了新的血腦屏障破壞機制[5]。各機制之間相互聯系,互為影響,錯綜復雜,尚無統一認識。目前臨床用藥品種少,主要有美金剛、多奈哌齊、石杉堿甲、卡巴拉汀、加蘭他敏五種藥物,且療效有限,對治療AD新藥的研制有迫切需求。結構決定性質,對于已上市藥物的結構研究可以更好地幫助我們了解藥物的本質。理論方法是銜接分子結構與實驗結果的橋梁,理論計算幫助我們從分子水平理解宏觀性質。這五種抗AD藥物在油性狀態下的結構和性質,系統的整體和局部反應性描述未見報道,在模擬大腦環境下的計算研究將更助于本課題組從分子水平上理解抗AD藥物的作用機制。
1 材料和方法
采用密度泛函理論(density functional theory,DFT)中雜化泛函B3LYP[6]方法,在TZVP基組水平上對五種藥物進行幾何結構優化和電子結構計算,在此基礎上獲得分子表面靜電勢(molecular electrostatic potential,MEP)、紫外-可見光譜(UV-Vis)和概念密度泛函系列指數。考慮到藥物作用大腦環境,采用環己烷為溶劑進行模擬,運用導體極化連續模型(conductor-like polarizable continuum model,CPCM)進行模擬計算,計算工作通過G16[7]程序包完成,圖像借助Gaussian View 6.0和Multiwfn3.6[8]軟件進行分析和繪制。
2 結果
2.1 幾何結構分析 將五種藥物在B3LYP/TZVP水平下優化,得到最穩定構象及部分鍵長見圖1。五種藥物均具有環狀疏水結構,如芐基、吡啶、苯環等,這與抗AD藥物需要通過血腦屏障,具有一定的親油性相吻合,可見抗AD藥物有一定的結構共性,而分子本身結構的差異,又導致了其作用機制的不同。觀察五種藥物分子極性鍵的情況,以美金剛C4-N13為基準,當C原子上連有苯環時,C-N鍵長稍有縮短,加蘭他敏C14-N15與多奈哌齊C17-N14鍵長均為1.459?。當C原子雙鍵連接O原子時,C-N鍵長縮短更加明顯,石杉堿甲C12-N13為1.400?,而卡巴拉汀C4-N3為1.354?,相較之下鍵長更短,鍵能更高。由于N原子上的孤對電子與苯環的π電子形成了共軛效應,使得兩個原子結合得更牢固,這種類似于肽鍵的結構印證了在N3-C4-O16之間存在共軛。朱維良等[9]等運用量子化學計算石杉堿甲單分子狀態的鍵長數據顯示C12-O14為1.217?,C2-N18為1.538?,N13-H29為0.997?,而形成石杉堿甲-乙酰膽堿酯酶(acetylcholine esterase,AChE)復合物后C12-O14為1.256?,C2-N18為1.477?,N13-H29為1.030?。我們在B3LYP/TZVP水平計算得到的數據與石杉堿甲單分子狀態進行比較,最大鍵長差為0.066?,與形成石杉堿甲-AChE復合物后比較,最大鍵長差為0.024?。計算數據與形成復合物后的數據更相近。陳衛民等[10]通過單晶X-射線測定了卡巴拉汀晶體結構,測得N13-C18為1.490?,N13-C12為1.525?,C6-O5為1.405?,C4-O5為1.360?,C4-O16為1.203?,與理論計算值相比,最大鍵長差為0.049?。RAVIKUMAR等[11]測得多奈哌齊晶體結構中C1-O25為1.364?,C2-O26為1.354?,C9-O24為1.220?,C8-C10為1.519?,C7-C8為1.537?,C10-C11為1.538?,C17-C18為1.500?,C17-N14為1.504?,與理論計算值相比,最大鍵長差為0.045?。理論數據和實驗晶體結構數據相近。
2.2 MEP分析 MEP是量子化學用于研究分子反應性、分子與受體相互作用及其他現象的重要工具。通過MEP能直觀感受到電子云分布狀態,預測分子親核親電反應活性位點。五種藥物在B3LYP/TZVP水平下的MEP見圖2,美金剛整體含一處負的靜電勢,分布在氮原子周圍,最小極值點亦在N13附近,其他部分呈正的靜電勢。加蘭他敏整體有3處含負的靜電勢,主要分布在氧原子和氮原子周圍。最小極值點在醚氧鍵和羥基附近,最大極值點在羥基附近。石杉堿甲整體有兩處含有負的靜電勢,分別在氧原子和氮原子上,最大極值點在N13附近而最小極值點在O14附近。多奈哌齊整體有三處含有負的靜電勢,為茚酮部分、芐基部分和甲氧基部分。最小極值點在O24附近,最大極值點在C3附近。卡巴拉汀整體有兩處含有負的靜電勢,分別在氮原子及氨基甲酸酯部分,最大極值點在N3附近,最小極值點在O16附近。擁有不同電性部位導致了五種藥物分子理化性質的差異。

圖1 五種抗AD藥物在B3LYP/TZVP水平下的幾何結構及部分鍵長數據(?)
有研究表明加蘭他敏的羥基能夠以氫鍵的形式與酶發生作用[12],多奈哌齊吡啶環中的N質子化后可以在AChE的通道部分產生陽離子-π作用,同時與水產生氫鍵。石杉堿甲羰基氧與AChE催化活性位點(catalytic active site,CAS)形成氫鍵,氨基上的氮原子在AChE的通道部分形成氫鍵。氫鍵本質是強極性鍵上的氫原子與帶有負電荷的原子之間的靜電作用力,對于生物高分子反應有著重要的意義。而對于富含芳香環氨基酸的AChE來說,這五種藥物也含有豐富的芳香環,因此發生在芳環之間的π-π堆積作用,與氫鍵一樣是重要的非共價鍵形式。研究表明加蘭他敏環己烯上的雙鍵與AChE的CAS位點形成π-π疊加[13],多奈哌齊芐基部分可與AChE的CAS位點產生π-π堆積,茚酮部分與AChE外周陰離子位點(peripheral anionic site,PAS)形成π-π堆積[14]。結合五種藥物的MEP不難發現電性差異決定了分子間作用力、作用位點的不同,電性極值點往往對應藥物與酶作用的位點,這對改變蛋白質構象具有關鍵性的作用,是反應核心。石杉堿甲的合成研究也驗證了這一點,天然提取的石杉堿甲構型為左旋的單一對映體,唐希燦等[15]將其對映異構體、外消旋體對AChE的抑制活性與之進行比較后認為右旋異構體活性和選擇性都大為降低是由于構型不匹配,環外亞乙基甲基的氫無法與酶形成有效氫鍵。

圖2 五種抗AD藥物在B3LYP/TZVP水平下的MEP圖
2.3 前線分子軌道分析 前線分子軌道(frontier molecular orbital,FMO)理論將分子周圍排布的電子云根據能量細分為不同能級的分子軌道,指出其中的最高占據軌道(highest occupied molecular orbital,HOMO)簡稱H,最低空軌道(lowest unoccupied molecular orbital,LUMO)簡稱L,是決定一個體系發生反應的關鍵。△ε(L-H)表示HOMO與LUMO的能差,反應了分子形成過渡態所需的能壘大小,差值越小,則能壘越小,越容易發生反應,化合物活性也就越高。
五種藥物的HOMO、LUMO及能差見表1,由表1可知,四種抗AChE藥物之間略有差異,但活性明顯高于N-甲基-D-天冬氨酸受體(N-methyl-D-aspartic acid receptor,NMDAR)拮抗劑美金剛。根據FMO,五種藥物可能的反應性順序為多奈哌齊>石杉堿甲>卡巴拉汀>加蘭他敏>美金剛。

表1 五種抗AD藥物分子的最高占據軌道和最低空軌道能及其能差
2.4 紫外-可見吸收光譜分析 五種藥物UV-Vis數據見表2,美金剛的UV-Vis存在兩個吸收峰,141.0 nm 處吸收強度大,躍遷類型屬于σ→σ*躍遷。美金剛化學結構獨特,分子結構中無共軛雙鍵,一般紫外-可見分光光度計只能提供190~850 nm范圍的單色光,因此在紫外區實驗無法觀測到,但是我們的理論計算能夠提供UV-Vis數據。其余四種抗AChE藥物的最大吸收峰落在179~208 nm之間,躍遷形式均為π→π*躍遷。李霞等[16]測得甲醇溶液中加蘭他敏在289nm處有最大吸收。王曉燕[17]對氫溴酸加蘭他敏標準品進行紫外掃描結果顯示,在322、254及289 nm處有較強吸收。LIU等[18]測得乙醇溶液中石杉堿甲在231和313 nm處有最大吸收。胡征[19]測得多奈哌齊在甲醇溶液中206.6 nm處有最大吸收,強度為0.921,在0.1 mol/L的HCl溶液中207.4 nm處有最大吸收,強度為1.212。王晗寧[20]測得溶劑為乙酸乙酯時,多奈哌齊在264和315 nm處分別有吸收,其中264 nm處有最強吸收峰。郭淼[21]測得甲醇溶液中卡巴拉汀最大吸收波長210 nm,且在262 nm處有一個特征吸收峰。比較可知隨溶液極性增加化合物吸收強度增大,整體發生紅移現象。

表2 五種藥物紫外吸收光譜數據
2.5 概念密度泛函分析 概念密度泛函理論適用于多電子體系結構的量子力學分析,是DFT的重要的分支,它把與化學相關的概念和原理從密度泛函中抽取出來,通過理論推導獲得計算參數,賦予了從前已知意義但是相對模糊的化學概念一個相對精確的描述。提出了全局反應性指數,包括:電離勢(IP)、電子親和勢(EA)、化學勢(μ)、電負性(χ)、整體硬度(η)、整體柔軟度(S)和整體親電指數(ω)。
五種藥物的全局反應性指數見表3。μ和η能體現分子的穩定狀態,五種藥物的η相差不大且皆為正值,表明對微干擾下化學體系電子云畸變的抵抗能力較強。多奈哌齊μ最低而加蘭他敏μ最高,表明多奈哌齊更穩定。多奈哌齊的χ和ω較大,說明其得電子能力強。S反映了分子周圍電子云分布情況,能體現化合物的反應活性,由S判斷藥物活性順序為多奈哌齊>石杉堿甲>加蘭他敏>卡巴拉汀>美金剛。鄭清川等[22]運用人類乙酰膽堿酯酶與抑制劑小分子對接后發現與AChE相互作用的強弱順序為多奈哌齊>石杉堿甲>卡巴拉汀,與我們由S判斷的活性順序相一致。在多奈哌齊、卡巴拉汀和加蘭他敏治療AD的安全性和耐受性比較研究中發現,多奈哌齊相較于其他兩種藥物胃腸道不良反應發生率更低,三種藥物中樞神經系統和心血管系統不良反應均較少發生,臨床應用多奈哌齊的依從性更好[23]。
在概念密度泛函中引入福井函數,以衡量當總電子數或外部化學勢發生變化時的電子密度差異。為進一步認識藥物分子活性情況對五種藥物進行了福井函數計算。結果發現,美金剛的親核部位具有偏向性,主要在N原子上連接氫原子的一側以及底部下方的位置,親電部位則不具偏向性,均勻分布在整個分子表面。加蘭他敏的親核部位同樣具有偏向性,主要分布于右下側苯環上,親電部位則偏向于右側苯環以及上方N原子部位。石杉堿甲的親核部位偏向于左邊的吡啶環上,親電部位則均勻分布于整體分子。多奈哌齊的親核部位主要位于左側茚酮結構上,親電部位主要在右邊吡啶環一側。卡巴拉汀親核部位主要在中間苯環上,親電部位主要在右邊氨基甲酸酯結構,見圖3。
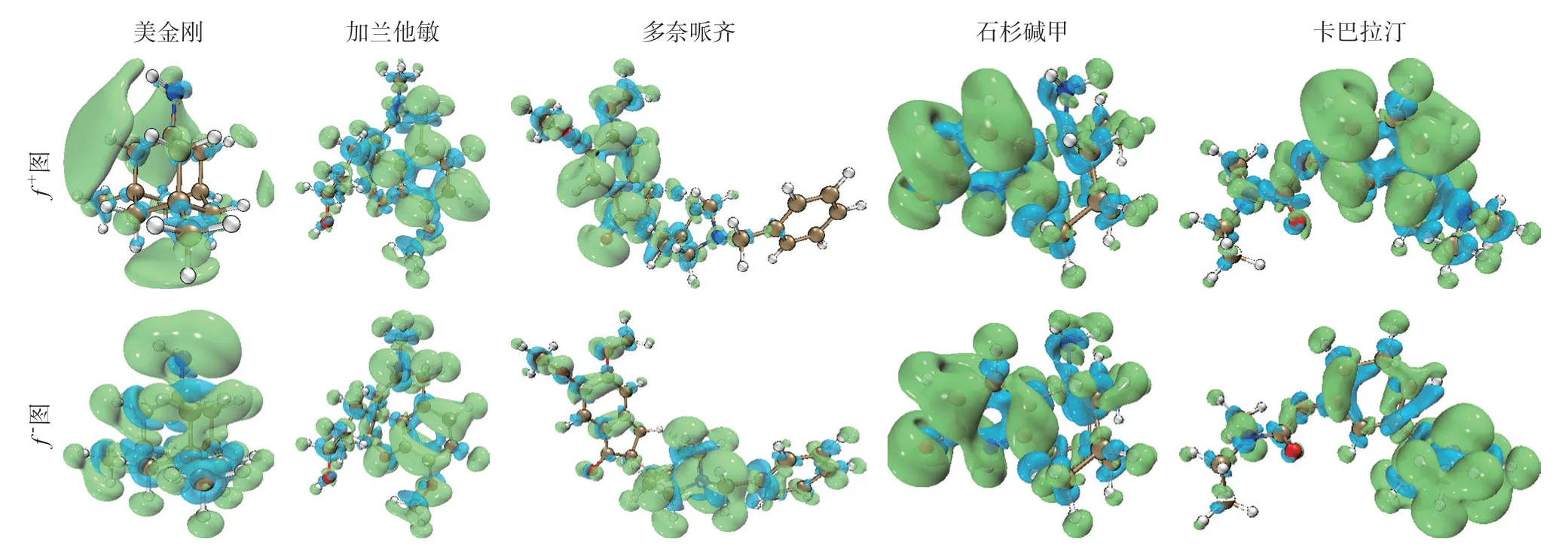
圖3 五種抗AD藥物的福井函數圖
五種藥物分子親電位置的差異與他們結構中是否帶有富余的電子密切相關。相對親電指數最大值分別為美金剛H14位4.730,加蘭他敏C5位6.391,石杉堿甲C10位2.923,多奈哌齊C9位23.071,卡巴拉汀C8位25.006。結合相對親電指數來看,多奈哌齊和卡巴拉汀親電能力強,美金剛、加蘭他敏和石杉堿甲親電能力較弱。親核位置的差異不僅與分子所帶電荷量相關,而且與分子結構的空間位置阻力也有著密切的聯系,相對親核指數最大值分別為美金剛N13位12.514,加蘭他敏N15位203.804,石杉堿甲C8位2.602,多奈哌齊N14位152.297,卡巴拉汀N13位86.031。結合相對親核指數來看,加蘭他敏、多奈哌齊、卡巴拉汀的親核能力強,美金剛和石杉堿甲親核能力較弱。
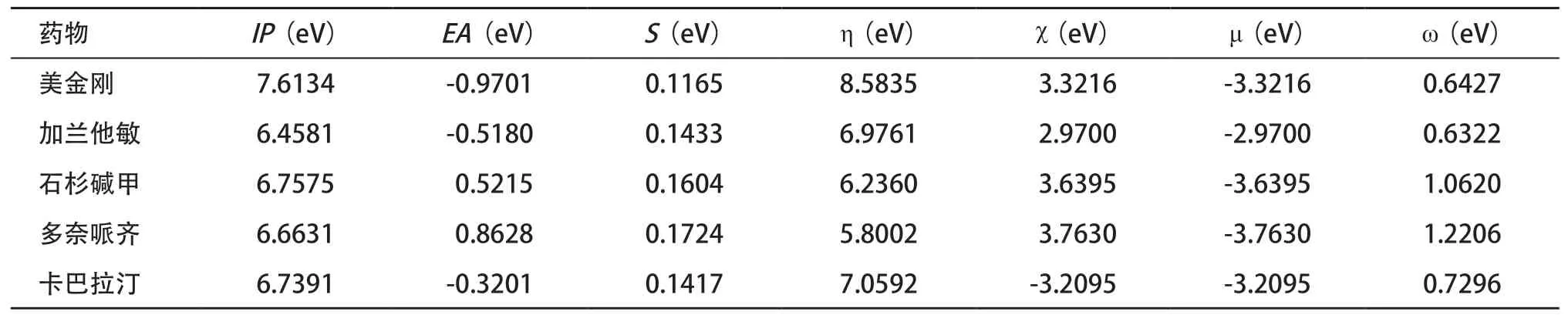
表3 五種抗AD藥物在B3LYP/TZVP水平下的全局反應性指數
3 討論
本研究在B3LYP/TZVP水平對美金剛、加蘭他敏、石杉堿甲、多奈哌齊和卡巴拉汀的分子結構、電子結構、UV-Vis以及概念密度泛函進行分析,結構分析比較出形成電子共軛效應使得極性鍵鍵長縮短。同時顯示計算數值與實驗數據吻合較好,表明借助DFT在B3LYP/TZVP水平可較好的研究抗AD藥物。電子結構分析顯示電性差異決定了分子間作用位點不同,特別是電性極值點,往往對應著與蛋白質形成非共價鍵的位置,是藥物作用的關鍵,這對今后抗AD藥物作用位點研究可以提供一定的指導作用。前線軌道分析顯示五種藥物中四種抗AChE藥物活性略有差異但明顯高于NMDAR拮抗劑,UV-Vis分析結果顯示溶劑效應使分子的吸收波長發生紅移且吸收強度增強。全局反應性指數表明多奈哌齊為五種抗AD藥物中軟度最大、電負性最大、親電指數最大、化學勢最低的藥物,藥理作用最強。由福井函數結合相對親電指數與相對親核指數得出多奈哌齊和卡巴拉汀親電、親核能力都很強,加蘭他敏親核能力強,美金剛和石杉堿甲親電、親核能力均比較弱。本研究結果為深入探索抗AD藥物作用機制以及藥物活性比較提供一定的計算支持。
