交聯(lián)改性對敷料用殼聚糖/聚氧化乙烯納米纖維膜性能的影響
2021-01-06 07:14:40汪希銘
紡織學(xué)報 2020年12期
汪希銘,程 鳳,高 晶,王 璐
(東華大學(xué) 紡織學(xué)院,上海 201620)
殼聚糖(CS)及其衍生物作為可再生天然高聚物,由于其獨(dú)特的理化性質(zhì)及廣泛的來源而受到持續(xù)地關(guān)注。CS具有優(yōu)異的組織相容性、生物可降解性、非致敏性、無細(xì)胞毒性以及獨(dú)特的陽離子抗菌性能[1],因此,其在藥物遞送、藥物緩釋[2]、組織工程[3]、傷口敷料[4]等領(lǐng)域被大量應(yīng)用。靜電紡絲是一種快速、高效地制備多尺度微納米纖維成形的手段。通過靜電紡成形的纖維膜由連續(xù)的超細(xì)纖維組成,具有高孔隙率、高透氣性、多尺度孔徑分布、高比表面積的特點(diǎn)[5],且具有與細(xì)胞外基質(zhì)相類似的納米纖維網(wǎng)絡(luò)結(jié)構(gòu)。
近年來,靜電紡殼聚糖納米纖維膜作為傷口敷料的研究不斷被報道。付譯鋆等[6]以環(huán)丙沙星為藥物模型,利用靜電紡絲方法獲得了具有載藥功能的CS/聚乙烯醇(PVA)納米纖維膜,其成形良好,具有可控且相對較低的藥物釋放速率。Adeli等[7]制備了靜電紡聚乙烯醇/CS/淀粉納米纖維膜,其在干濕狀態(tài)下均表現(xiàn)出良好的力學(xué)性能、高孔隙率、高吸濕率和較高的水蒸氣透過率,可有效地保護(hù)傷口免受細(xì)菌感染。Chen等[8]利用序列靜電紡絲法制備了CS/PVA多層靜電紡絲膜,其優(yōu)化的空間結(jié)構(gòu)能夠?yàn)榧?xì)胞黏附和遷移提供適宜的微環(huán)境,并表現(xiàn)出優(yōu)異的生物相容性、抗菌活性以及促再生功能。Ren等[9]將負(fù)載廣譜抗菌藥物氯己定二葡萄糖酸酯的埃洛石納米管(HNTs)分散至絲素蛋白(SF)/CS 混合物中,通過靜電紡獲得具有微納結(jié)構(gòu)的纖維膜敷料,該纖維膜兼具穩(wěn)定的力學(xué)性能、抗菌活性和止血性能,較一般載藥敷料具有更長的藥物釋放時間,且具備酸性環(huán)境下藥物響應(yīng)性釋放的功能,在生物醫(yī)學(xué)方面有很大的應(yīng)用潛力。
由于CS具有特殊的剛性分子鏈結(jié)構(gòu)及聚陽離子特性,純CS很難被溶解而紡制成納米纖維膜[10],因此,一般將CS與其他高聚物如聚環(huán)氧乙烷(PEO)[11]或聚乙烯醇(PVA)[12]共混并溶于稀酸溶液[13]。共混物之間所形成的氫鍵導(dǎo)致高聚物之間的分子鏈纏結(jié)作用增大,降低紡絲溶液的導(dǎo)電性,從而大大地改善殼聚糖的可紡性[14]。此外,CS基納米纖維在水相環(huán)境中會吸濕溶脹,降低纖維膜的孔隙率、透氣性,且對其獨(dú)特結(jié)構(gòu)優(yōu)勢造成破壞,因此,作為傷口敷料功能層,CS基纖維膜必須通過交聯(lián)改性處理,以獲得更好的結(jié)構(gòu)穩(wěn)定性和耐水性,解決其在傷口組織液浸潤后的結(jié)構(gòu)不穩(wěn)定問題。本文通過化學(xué)交聯(lián)劑戊二醛(GA),對CS/PEO靜電紡納米纖維膜進(jìn)行交聯(lián)處理,并通過掃描電子顯微鏡(SEM)、 X射線衍射儀(XRD)、紅外光譜儀(FT-IR)、 單纖維強(qiáng)力儀等對交聯(lián)前后CS/PEO納米纖維膜的微觀形貌、結(jié)構(gòu)穩(wěn)定性和力學(xué)性能等進(jìn)行表征,探究交聯(lián)改性對CS/PEO納米纖維膜結(jié)構(gòu)和性能的影響,以期更好地應(yīng)用于傷口敷料。
1 實(shí)驗(yàn)部分
1.1 實(shí)驗(yàn)材料
殼聚糖(CS,脫乙酰度大于92%,黏度為180 mPa·s,浙江金殼生物化學(xué)有限公司);聚環(huán)氧乙烷(PEO,平均分子質(zhì)量為9×105ku,百靈威科技有限公司);GA溶液(分析純,質(zhì)量分?jǐn)?shù)為25%,國藥集團(tuán)化學(xué)試劑有限公司)。
1.2 實(shí)驗(yàn)儀器
SS-1型靜電紡絲機(jī),北京永康樂業(yè)科技發(fā)展有限公司;LLY-006型單纖維強(qiáng)力儀,萊州市電子儀器有限公司;TM-3000型掃描電子顯微鏡,日本Hitachi公司;Nicolet 6700型傅里葉變換紅外光譜儀,美國 Thermo Fisher公司;D/max-2550 PC型X射線多晶衍射儀,日本Rigaku公司。
1.3 實(shí)驗(yàn)方法
1.3.1 CS/PEO紡絲溶液的制備
室溫條件下,使用90%的冰醋酸溶液作為溶劑,將CS與PEO以質(zhì)量比為9∶1,經(jīng)磁力攪拌至完全溶解,得到質(zhì)量分?jǐn)?shù)為5%的CS/PEO紡絲溶液。
1.3.2 CS/PEO納米纖維膜的制備
取5 mL注射器吸取一定量的CS/PEO紡絲溶液后,將其頭端與內(nèi)徑為1.7 mm的針頭相連,開啟電源進(jìn)行靜電紡絲。電壓設(shè)置為10 kV,推注速度為0.8 mL/h。 針頭到接收裝置的距離為20 cm。連續(xù)紡絲6 h后制得CS/PEO靜電紡納米纖維膜,真空干燥4 h后置于干燥器中待用。
1.3.3 CS/PEO納米纖維膜的交聯(lián)改性
取一定量的GA溶液于培養(yǎng)皿中,放置于干燥器底部,將制備好的CS/PEO纖維膜裁剪為合適大小后,放置在干燥器底部的支架上,于室溫下利用GA蒸汽進(jìn)行交聯(lián),時間分別為0(未交聯(lián))、4、8、12和24 h。交聯(lián)完成后試樣經(jīng)真空干燥處理一定時間,使殘留GA 完全去除后密封保存于干燥器中。
1.3.4 微觀形貌觀察
通過掃描電子顯微鏡對交聯(lián)處理前后纖維的微觀結(jié)構(gòu)進(jìn)行觀察,從不同圖像中選取至少100個點(diǎn)對纖維直徑進(jìn)行測量分析。加速電壓設(shè)置為5 kV。
1.3.5 化學(xué)結(jié)構(gòu)測試
通過傅里葉變換紅外光譜儀表征CS粉末、PEO纖維膜、CS/PEO納米纖維膜的化學(xué)結(jié)構(gòu),波數(shù)范圍為4 000~400 cm-1。
1.3.6 結(jié)晶結(jié)構(gòu)測試
通過X射線多晶衍射儀測定各樣品的結(jié)晶結(jié)構(gòu)。參數(shù)設(shè)置為:電壓40 kV,掃描速度3 (°)/min,掃描范圍5°~60°。
1.3.7 吸水率和質(zhì)量損失率測試
將5種交聯(lián)時間(0、4、8、12、24 h)處理后的樣品,采用稱取質(zhì)量法測量其吸水率及質(zhì)量損失率。將樣品裁剪成一定大小,記錄其初始干態(tài)質(zhì)量(m0);然后置于模擬PBS緩沖液(pH=7.2)中24 h后,使用蒸餾水沖洗3次,將濾紙置于試樣表面以吸干其表面水分,并記錄濕態(tài)質(zhì)量(mw),冷凍后真空干燥并記錄纖維膜質(zhì)量(md)。3次平行實(shí)驗(yàn)后,由下式求得其平均吸水率(W)和平均質(zhì)量損失率(S):
1.3.8 力學(xué)性能測試
采用電子單纖維強(qiáng)力儀在溫度為20 ℃、相對濕度為 65%條件下,以10 mm/min的拉伸速度對試樣進(jìn)行拉伸實(shí)驗(yàn),隔距長度設(shè)置為 10 mm。每種試樣測5次,取平均值。
2 結(jié)果與討論
2.1 交聯(lián)改性對納米纖維膜形貌的影響
殼聚糖基納米纖維膜在傷口組織液浸潤后需保持細(xì)胞外基質(zhì)的類似結(jié)構(gòu),因此,對其進(jìn)行交聯(lián)改性后的纖維形貌進(jìn)行觀察,結(jié)果如圖1所示。圖中左側(cè)為CS/PEO納米纖維膜分別經(jīng)25%的GA蒸汽于室溫下交聯(lián)0、4、8、12和24 h的微觀形貌圖。可知,不同交聯(lián)時間的纖維直徑分別由未交聯(lián)的310 nm 增加至404、436、537和598 nm。這可能是由于在交聯(lián)反應(yīng)過程中,纖維受到GA蒸汽中水分的影響而發(fā)生溶脹效應(yīng),隨交聯(lián)時間的增加,纖維與水的作用時間變長,纖維溶脹程度不斷增加,體現(xiàn)為纖維直徑變大。
圖1中右側(cè)為交聯(lián)處理前后纖維膜在PBS中浸泡24 h后的微觀形貌。可以看出,未交聯(lián)的纖維膜在PBS浸泡后,發(fā)生明顯地溶脹、軟化和交纏;而在交聯(lián)后,隨著交聯(lián)處理時間不斷增加,經(jīng)PBS浸泡后纖維形貌均勻性得到提升,光滑度不斷提高,這說明交聯(lián)處理可有效提高CS/PEO納米纖維膜的耐水性,且交聯(lián)時間越長,耐水性越好,這為其抵抗傷口組織液浸入發(fā)生結(jié)構(gòu)形變提供了很好的解決方案。

圖1 不同交聯(lián)時間的納米纖維膜經(jīng)PBS浸泡24 h 前后的掃描電鏡照片(×5 000)Fig.1 SEM images of nanofiber membranes under different cross-linking time before and after soaked in PBS for 24 h(×5 000)
2.2 交聯(lián)改性對納米纖維膜結(jié)構(gòu)的影響
圖2示出交聯(lián)改性前后納米纖維膜紅外光譜圖。可知,加入PEO后 3 400~3 100 cm-1處醇羥基(—OH)、 氨基(—NH)吸收峰在殼聚糖分子內(nèi)氫鍵的作用下重疊增寬,與1 594 cm-1處CS上氨基(—NH)的吸收峰[15-16]分別轉(zhuǎn)移到3 362和1 558 cm-1處,且2 876 cm-1處—CH2的伸縮振動吸收峰強(qiáng)度增強(qiáng)。這是由于CS在PEO中醚鍵的影響下,原有的分子內(nèi)氫鍵轉(zhuǎn)變?yōu)榘被cPEO中醚鍵形成的分子間氫鍵[17],使得CS結(jié)晶度降低從而提升了其可紡性。與純CS相比,CS/PEO共混纖維膜中—CH2吸收峰峰強(qiáng)增加大且向高波數(shù)移動;且純CS紅外光譜圖中位于1 070~1 030 cm-1之間的2個孤峰變?yōu)椴豢煞值募绶澹⑾蚋卟〝?shù)移動[15]。由此推斷出PEO加入后,CS分子中的分子內(nèi)氫鍵被破壞,CS中的氨基、酰胺基與PEO中的氫分別產(chǎn)生了新的氫鍵作用,影響了CS大分子的結(jié)晶結(jié)構(gòu),有效提升了CS的紡絲性能,且證明了PEO與CS相容性良好[18-19]。
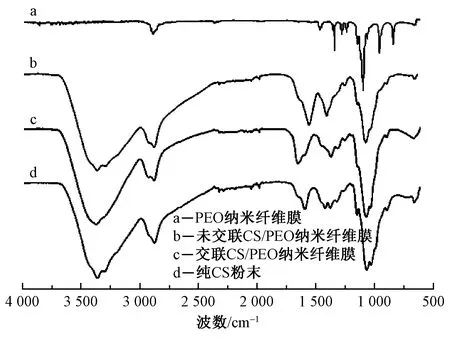
圖2 納米纖維膜和CS的紅外光譜圖Fig.2 FT-IR spectra analysis on nanofiber membranes and CS

圖3示出納米纖維膜的X射線衍射光譜圖。可知,在純CS粉末的X射線衍射圖中,特征衍射峰分別位于2θ為14.5°、20.1°、28.1°以及29.5°處,其結(jié)晶度為44.72%。
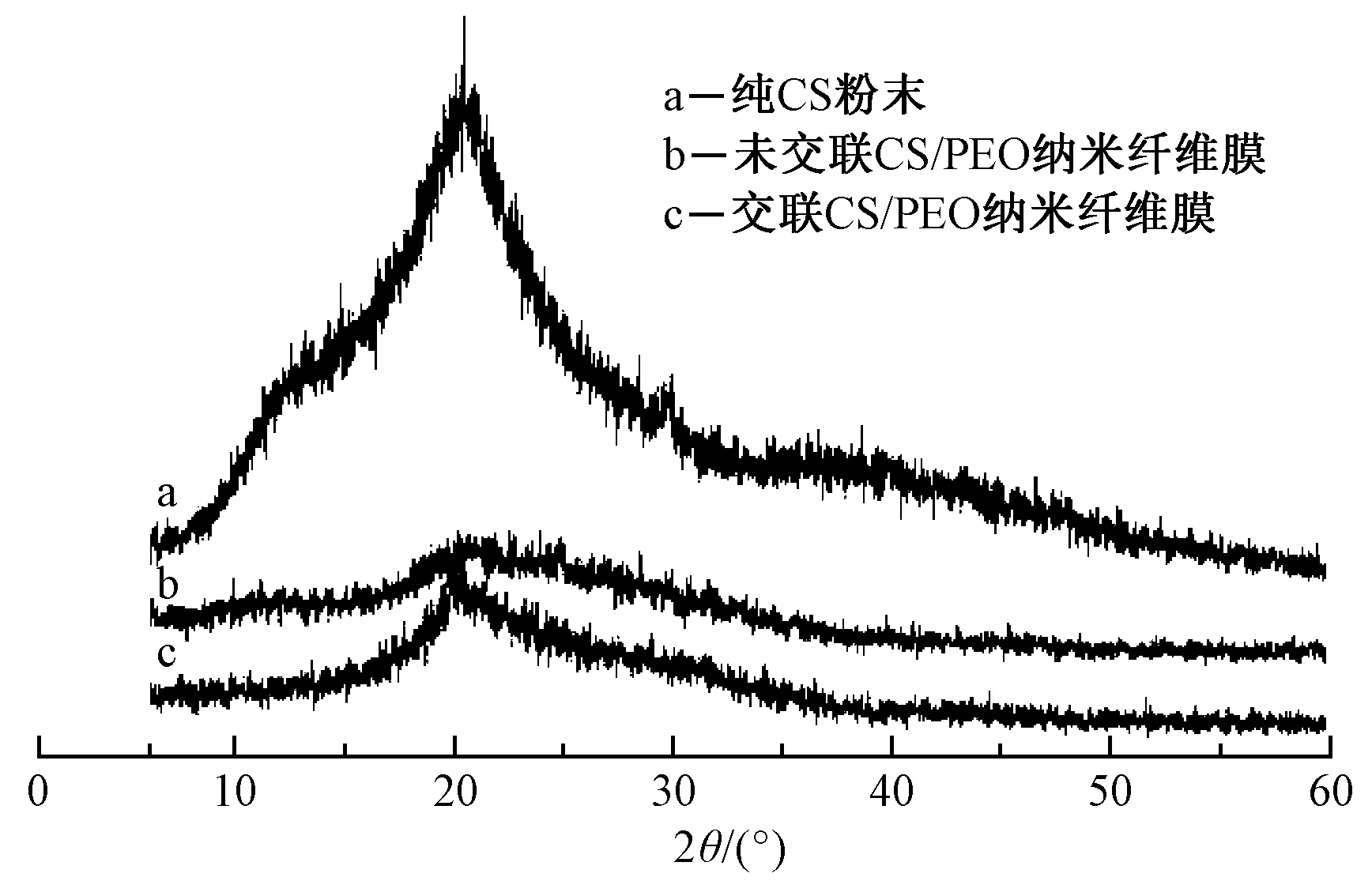
圖3 納米纖維膜和CS的X射線衍射圖Fig.3 X-ray diffraction spectra of nanofiber membranes and CS
2.3 交聯(lián)改性對吸水率和質(zhì)量損失率影響
圖4示出不同交聯(lián)時間的CS/PEO納米纖維膜在PBS中處理24 h后的吸水率和質(zhì)量損失率。
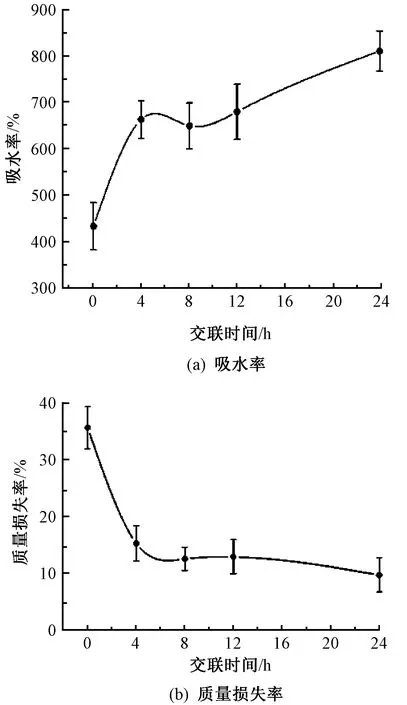
圖4 不同交聯(lián)時間的納米纖維膜在PBS溶 液中浸泡24 h后的吸水率與質(zhì)量損失率Fig.4 Water absorption(a) and dissolution(b) ratio of nanofiber membranes under different cross-linking time after soaked in PBS for 24 h
從圖4 (a)可見,交聯(lián)處理4 h后,隨著交聯(lián)時間的不斷增加,纖維膜吸水率呈現(xiàn)先降低后升高的趨勢。由圖4 (b)可知,交聯(lián)改性后的納米纖維膜質(zhì)量損失率大幅度降低,且隨著交聯(lián)改性時間的增加,纖維膜的質(zhì)量損失率呈小幅度下降趨勢。其原因可能是交聯(lián)處理后,CS分子鏈之間形成共價交聯(lián)網(wǎng)絡(luò),纖維膜結(jié)構(gòu)變得更加緊密,阻止了PEO的溶出,表現(xiàn)為纖維膜的質(zhì)量損失率大幅度下降,而吸水率在排除纖維膜溶失部分影響后有所降低;與此同時,交聯(lián)處理后CS分子鏈中部分氨基脫離分子間氫鍵的束縛,轉(zhuǎn)變?yōu)橛坞x態(tài),從而造成了纖維膜吸水性的提高;隨著交聯(lián)時間的繼續(xù)延長,過度交聯(lián)使得纖維間粘合破裂,纖維中孔隙增多,因而吸水率不斷增加。可見,適當(dāng)時間交聯(lián)處理后的納米纖維膜的吸水率和質(zhì)量損失率變化特點(diǎn)符合作為傷口敷料功能層的實(shí)際需求,即交聯(lián)處理后的納米纖維膜能夠更多地吸收組織液,以達(dá)到清創(chuàng)的效果;同時內(nèi)部結(jié)構(gòu)更加穩(wěn)定,自身溶失較少,不會對敷料結(jié)構(gòu)產(chǎn)生較大的影響。
2.4 交聯(lián)改性對納米纖維膜力學(xué)性能影響
表1示出CS/PEO納米纖維膜在干燥狀態(tài)下的拉伸性能測試結(jié)果。可知:隨交聯(lián)時間的不斷增加,CS/PEO納米纖維膜的斷裂強(qiáng)度呈先升高后降低趨勢,在交聯(lián)處理時間為8 h時斷裂強(qiáng)度與斷裂伸長率均達(dá)到最大值,分別為(3.85±0.59) MPa與(4.62±1.24)%。這是由于CS分子中的氨基與GA分子中的醛基發(fā)生反應(yīng)形成剛性亞胺鍵,從而使主要由氫鍵、范德華力等作用力形成的較弱自連網(wǎng)絡(luò),被共價鍵形成的網(wǎng)絡(luò)所取代,因此,納米纖維膜的強(qiáng)度和脆性均得到顯著提升。由于納米纖維膜作為傷口敷料的功能層覆蓋于傷口之上,對其拉伸和撕扯的力較弱,因此,納米纖維膜力學(xué)性能一定程度的變化不會對其使用產(chǎn)生過度影響。
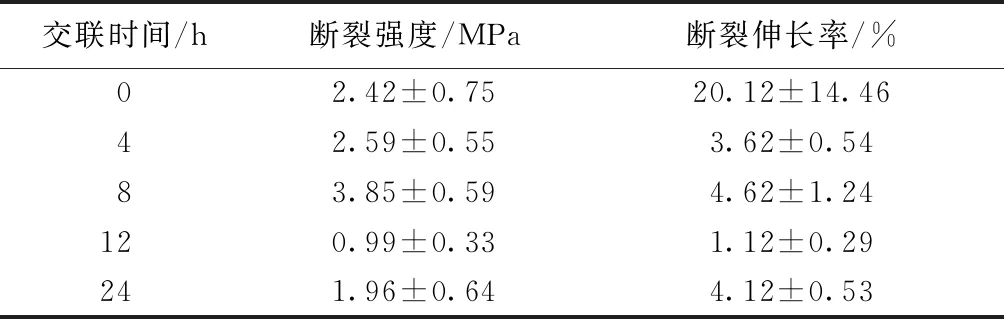
表1 不同交聯(lián)時間的納米纖維膜的力學(xué)性能Tab.1 Mechanical property of nanofiber membranes under different cross-linking time
3 結(jié) 論
本文采用乙酸為溶劑制備殼聚糖/聚氧化乙烯(CS/PEO) 靜電紡納米纖維膜,探究戊二醛交聯(lián)不同時間后纖維膜微觀形態(tài)、結(jié)構(gòu)穩(wěn)定性及力學(xué)性能等的變化。CS/PEO納米纖維膜交聯(lián)前后的掃描電鏡測試表明,隨著交聯(lián)時間的不斷增加,纖維在水的作用下發(fā)生溶脹,直徑有所增加;納米纖維膜PBS浸泡實(shí)驗(yàn)顯示,隨著交聯(lián)時間的延長,PBS處理后纖維的形貌更加穩(wěn)定,光滑度逐漸增加,交聯(lián)改性可顯著提高纖維膜的耐水性能,使其更適合應(yīng)用于傷口敷料。經(jīng)交聯(lián)處理后CS/PEO纖維膜中CS與GA分子生成剛性亞胺鍵,分子間形成共價交聯(lián)網(wǎng)絡(luò),并使得CS大分子固有結(jié)晶結(jié)構(gòu)發(fā)生重排。
CS/PEO纖維膜交聯(lián)前后的吸水率和質(zhì)量損失率測試結(jié)果表明,隨著交聯(lián)時間的不斷增加,纖維膜結(jié)構(gòu)致密化,其吸水率得到提升,質(zhì)量損失率降低,證明了交聯(lián)處理對纖維膜結(jié)構(gòu)穩(wěn)定具有積極影響;纖維膜的力學(xué)性能測試分析結(jié)果顯示,交聯(lián)處理后CS/PEO纖維膜隨著處理時間的增加其力學(xué)強(qiáng)度呈先升后下降趨勢,因此,適當(dāng)?shù)慕宦?lián)處理可一定程度上提升納米纖維膜的強(qiáng)度和韌性。
猜你喜歡
中學(xué)生數(shù)理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學(xué)生數(shù)理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
中國塑料(2016年12期)2016-06-15 20:30:07
汽車觀察(2016年3期)2016-02-28 13:16:26
食品界(2016年4期)2016-02-27 07:36:46
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
中國塑料(2015年4期)2015-10-14 01:09:19
應(yīng)用化工(2014年7期)2014-08-09 09:20:21
