基于PCR的稀有突變檢測方法
2021-01-07 10:23:44張絢張宏剛
生物化工 2020年6期
張絢,張宏剛
(1.國家知識產(chǎn)權(quán)局專利局專利審查協(xié)作廣東中心,廣東廣州 510500;2.廣州格拉姆生物科技有限公司,廣東廣州 510530)
基因突變是指由于DNA分子中堿基對的增添、缺失或改變而引起的基因結(jié)構(gòu)的改變。稀有突變則指存在大量的野生型背景的突變模板,即樣品基因組的突變比例非常低[1]。目前,應用分子生物學方法檢測患者體內(nèi)遺傳物質(zhì)的結(jié)構(gòu)和表達水平的變化的分子診斷技術(shù)在疾病的診斷和治療中占據(jù)舉足輕重的地位,而稀有突變的分子診斷作為基因突變檢測的一個重要組成部分,在技術(shù)的建立到臨床的應用過程中都面臨著巨大的挑戰(zhàn)[2]。
分子診斷中的很多領域都涉及了稀有突變的檢測,主要包括以下3個方面。(1)一些稀有突變可作為癌癥的早期診斷和預后標記物,這種檢測常以組織切片、體液(如血漿或血清)、尿液或糞便為模板(基因組DNA或RNA)來源的進行突變檢測。(2)病人的微小殘留病的檢測,微小殘留病指癌癥患者經(jīng)過治療并達到緩解后殘留的腫瘤細胞,可用來監(jiān)控病人的病情,預測病人的復發(fā)風險。(3)稀有突變的檢測在疾病的分級應用中也占有一席之地。
稀有突變檢測技術(shù)面臨的一個最大的難題是在野生型模板量大大超出了要檢出的突變模板量的情況下,如何能夠盡量減小野生型模板的影響,從而高靈敏度地將突變模板篩查出來。目前的PCR檢測技術(shù)為了解決這個難題,往往采用各種手段壓制野生型模板的擴增,同時富集突變模板,使突變模板的濃度比例增高而易于檢測。而在檢測方法的選擇上,需要根據(jù)實際情況,在靈敏度(即能檢測到的最低突變比例)和準確性間找到平衡點,此外,還要考慮所選方法的簡便性和成本。
基于PCR的稀有突變富集檢測方法主要有兩類,一類只能對已知突變進行富集檢測,另一類則還能對未知突變進行富集檢測。表1依據(jù)靈敏度的高低和富集對象的類型列出了較常見的一些基于PCR的檢測方法。接下來將針對其中比較常用的方法進行介紹。
1 針對已知突變的中等靈敏度檢測方法
目前,最常用的是ARMS和PNA或LNA介導的PCR。
1.1 ARMS
擴增阻礙突變系統(tǒng)技術(shù)[1-2]又稱等位基因特異性PCR,在突變檢測中應用很廣泛。該檢測方法基于TaqDNA聚合酶缺乏3'到5'外切校正活性,從而在ARMS引物的3'端設計等位基因特異性的堿基,有時為了提高特異性,還在引物的3'末端根據(jù)需要引入第二、第三個錯配堿基。當引物結(jié)合于模板時,由于該ARMS引物與野生型模板和突變型模板的錯配程度不同,而導致不匹配的模板的PCR延伸受阻,特異性地擴增匹配型模板。此技術(shù)與實時熒光PCR技術(shù)相結(jié)合,可以實現(xiàn)DNA突變的快速、準確、高通量檢測。
用ARMS引物檢測基因突變,建立體系時需全盤考慮,仔細優(yōu)化多個反應條件,但最核心的部分是ARMS引物的設計與篩選。ARMS引物設計時需融入多種考慮因素,引物設計軟件選擇出的引物和分值往往和實際情況有偏差,也不能估算不完全匹配引物對模板的擴增能力,所以對于突變點需進行較多的引物篩選。在ARMS引物3'端引入錯配時,考慮到不同突變類型序列不同,根據(jù)各個突變的序列特點,在3'端1、2、3位引入1~3個錯配堿基,使所設計引物3'端與野生型匹配度最差,與待檢測突變類型匹配度較高,然后在實驗過程中需不斷根據(jù)實驗結(jié)果分析選擇和調(diào)整引物。
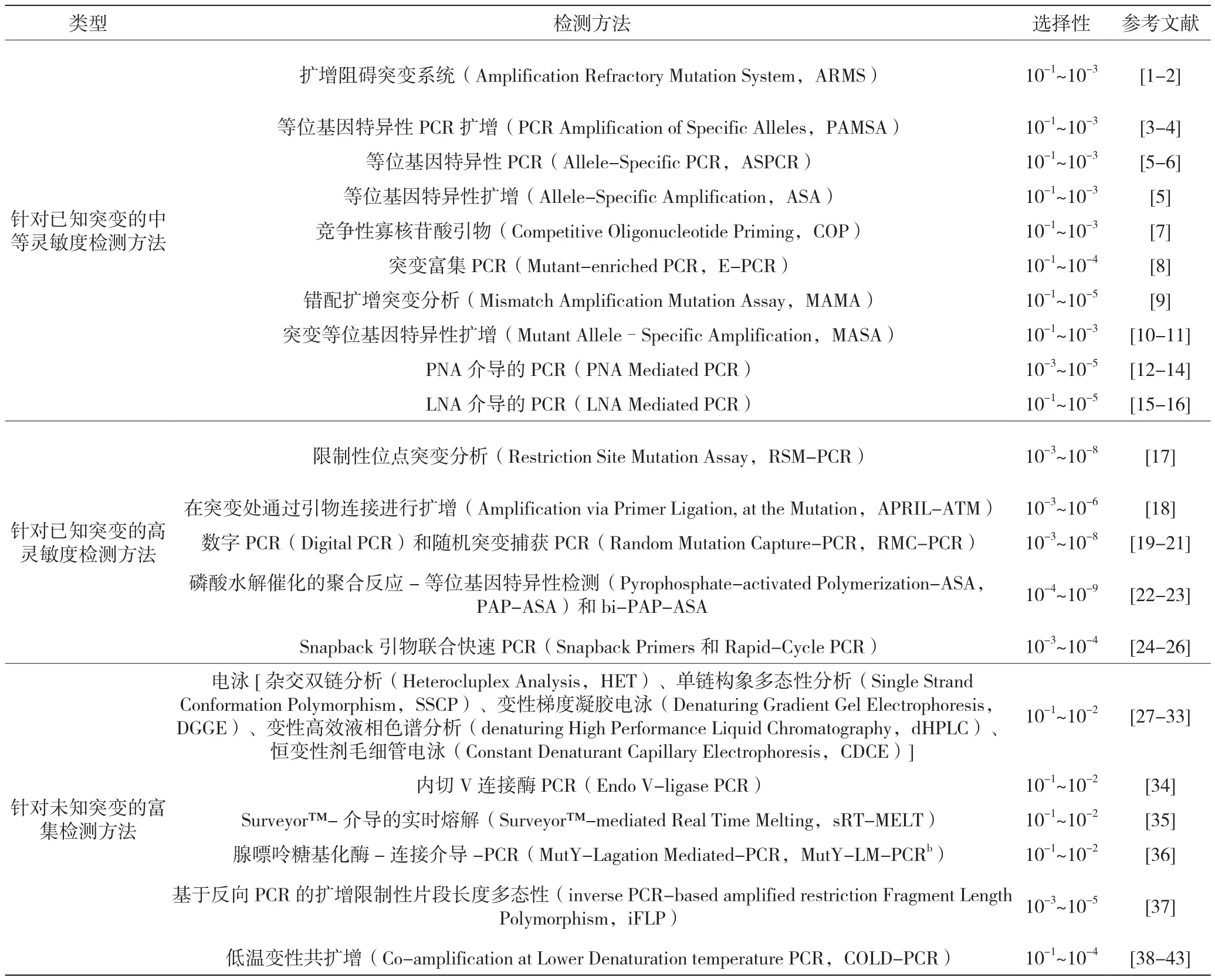
表1 稀有突變檢測方法
1.2 PNA或LNA介導的PCR
肽核酸(Peptide nucleic acid,PNA)是具有類多肽骨架的DNA類似物,PNA的主鏈骨架是由N(2-氨基乙基)-甘氨酸與核酸堿基通過亞甲基羰基連接而成的。鎖核酸(Locked nucleic acid,LNA)是一種經(jīng)過修飾的RNA,β-D-呋喃核糖的2’-O、4'-C位通過縮水作用形成剛性結(jié)構(gòu)。PNA和LNA都具有如下特點:(1)可以特異性地與DNA或RNA雜交,形成比ssDNA或ssRNA更穩(wěn)定的復合體;(2)DNA聚合酶對其沒有延伸活性;(3)在PCR反應過程中不會被DNA聚合酶酶切。正是基于這些特點,PNA[12-14]和LNA[15-16]常用于PCR反應中鎖住野生型模板的“夾子”,以避免野生型模板的擴增,從而使突變模板濃度升高而易于檢測。
2 針對已知突變的高靈敏度檢測方法
2.1 數(shù)字PCR(Digital PCR)和PAP-ASA
數(shù)字PCR[19-21]利用核酸樣品做模板,混合成完整的PCR體系后,將數(shù)微升的PCR反應體系隨機分布到將近1 000個納米級的反應倉(Reaction Chamber)中去。當起始的核酸模板濃度足夠低時,單分子核酸就會出現(xiàn)在反應倉格子中,對PCR產(chǎn)生熒光信號的格子進行計數(shù),從而達到對所研究的基因進行檢測或定量。該技術(shù)在基因突變檢測、拷貝數(shù)變化檢測及基因分型中應用較為廣泛。
PAP-ASA技術(shù)是近幾年發(fā)展起來的一種稀有突變檢測技術(shù),該技術(shù)利用了Taq聚合酶的焦磷酸水解活性和聚合活性。與普通引物不同的是,PAP引物的3'末端采用了ddNTP修飾,當該ddNTP與模板匹配時,Taq聚合酶的焦磷酸水解活性將其水解去除,從而恢復PAP引物的延伸能力;當該ddNTP與模板不匹配時,焦磷酸不被水解,從而無法延伸。焦磷酸水解活性的特異性和聚合酶活性本身的特異性相互疊加形成了PAP反應無可比擬的高特異性,靈敏度理論上可達10-9。目前,PAP的定性檢測通常采用先擴增后電泳分析結(jié)果的方法,不僅需要較長的時間和較大的勞動強度,而且PCR產(chǎn)物的后處理很容易導致樣品間的交叉污染,這對于稀有突變檢測來說是致命的。Song等人[23]將PAP技術(shù)整合到實時PCR檢測平臺,將擴增和檢測合二為一,不需要任何后操作,有效杜絕了產(chǎn)物污染,還具有卓越的定量能力。
然而,由于PAP反應涉及酶的兩次反應,所以需要較長的反應時間和較緩慢的升溫,運行這類程序?qū)崟rPCR儀有一定的損耗。
2.2 Snapback Primers and Rapid-Cycle PCR
Snapback引物聯(lián)合快速PCR[24-26]方法設計了一種Snapback引物,該引物包括兩部分,延伸部分(3′端)和尾巴部分(5'端)。延伸部分(3'端)為普通PCR引物;尾巴部分(5'端)與目的基因發(fā)生突變的區(qū)域互補,突變堿基所在位置位于此尾巴的中間。這樣做的目的是,當該引物以野生型序列為模板,擴增出野生單鏈時,該單鏈5'端由于互補序列的存在,將會形成一個穩(wěn)定的手柄結(jié)構(gòu),從而阻抑聚合酶的進一步擴增。當該引物以突變序列為模板進行擴增時,擴增出的產(chǎn)物DNA雖也可形成手柄結(jié)構(gòu),但由于突變的存在,該手柄結(jié)構(gòu)極不穩(wěn)定,易打開而發(fā)生聚合,由此便對突變模板進行富集。另外,Snapback引物的5′端還需引入兩個無關(guān)堿基來阻止形成手柄結(jié)構(gòu)后的自引物延伸作用。這種方法需要聯(lián)合快速循環(huán)PCR,需要變溫速率較高的PCR儀。
3 針對未知突變的富集檢測方法
針對未知突變的檢測方法往往涉及很多種酶的使用,增加了檢測方法的復雜性和操作時間。但是COLD-PCR[38-43]只通過改變擴增的程序即可達到對突變模板的富集。
COLD-PCR的基本原理是:當模板DNA中有突變存在時,擴增產(chǎn)物會形成兩類DNA雙鏈:同源二聚體雙鏈和異源二聚體雙鏈。這時,可通過降低退火溫度(Tc),選擇性地將含有突變的異源二聚體雙鏈打開,進一步進行擴增延伸,從而達到突變富集的作用。這里的退火溫度是COLD-PCR中最關(guān)鍵的因素,關(guān)系到整個COLD-PCR的成功與否,需要對其進行非常細致的優(yōu)化。此外,為了增大同源二聚體雙鏈和異源二聚體雙鏈之間穩(wěn)定性的差異,PCR產(chǎn)物不宜太長,一般200 bp以內(nèi)為佳。
COLD-PCR有兩類,一種是完全COLD-PCR(Full COLD-PCR),另一種是快速COLD-PCR(Fast COLD-PCR)。前者可對所有的突變類型進行富集,后者則只對發(fā)生突變后穩(wěn)定性下降,DNA解鏈溫度(Tm)下降的DNA鏈進行富集。因不同的需要可選用不同的類型。在都適用的情況下,后者的富集效果往往好于前者。
此外,發(fā)明COLD-PCR的作者還分析了兩類COLD-PCR對不同突變類型的富集程度。如C-G到T-A或A-T突變,完全COLD-PCR可富集5~12倍,快速COLD-PCR可富集10~100倍;T-A到A-T或C-G到G-C的突變,完全COLD-PCR可分別富集5~8倍和3~5倍;對于插入和缺失突變,完全COLD-PCR可富集50倍以上,快速COLD-PCR可富集100倍以上。
4 結(jié)語
稀有突變是突變檢測的一個重要組成部分,也是目前臨床應用上面臨的一個巨大挑戰(zhàn)。目前,稀有突變的檢測方法很多,但方法的高靈敏度往往意味著其復雜性。而且,有些方法也具有需昂貴儀器、費時或檢測成本高等缺點。此外,隨著人們對稀有突變的生物和臨床特性研究的深入,針對稀有突變的檢測技術(shù)也必會隨之不斷發(fā)展。
猜你喜歡
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產(chǎn)業(yè)(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
