鋰離子電池合金負極材料研究進展
2021-01-21 07:51:08馬家賓陳立坤賀艷兵
太原理工大學學報 2021年1期
關鍵詞:界面
彪 捷,馬家賓,陳立坤,賀艷兵
(清華大學 a.深圳國際研究生院,廣東 深圳 518055,b.材料學院,北京 100084)
鋰離子二次電池以其高能量密度在能量儲存技術中越來越重要,自1991年進入市場后,已經廣泛應用于便攜式電子設備、新能源汽車等領域,但是目前傳統鋰離子電池仍難以滿足對高能量密度儲能設備不斷增加的需求[1]。隨著大規模能源存儲需求的不斷增長,鋰金屬電池(LMBs)為電動汽車和智能電網的應用提供了一個能量密度的重大飛躍[2]。鋰金屬具有最負的電化學勢(-3.04 V vs. NHE)、較高的理論比容量(3 860 mAh/g)和極低的密度(0.534 g/cm3),被認為是鋰電池的“圣杯”。然而,由于在鋰金屬沉積/剝離的循環過程中,電沉積控制不佳和體積膨脹,導致鋰金屬的結構疏松、枝晶生長和“死鋰”的產生,同時鋰與電解液具有強烈的副反應,導致鋰金屬的應用一直受到阻礙[2]。合金負極以接近鋰金屬負極的高理論比容量和低電極電勢,成為極具潛力的下一代鋰離子電池負極材料。合金是由兩種或兩種以上的金屬或金屬與非金屬經一定方法所合成的具有金屬特性的物質,可以在電勢差的驅動下進行脫嵌鋰反應,鋰原子從原始晶格中脫嵌形成新的混合固溶相。根據組成元素的數目,鋰合金負極可分為二元合金負極(Li-M,M=Mg、Ag、Zn、C、Si、Sb、Bi等)和多組元合金負極(Li-X-M,X=Si、Sn,M=Mg、Ag、C等)[3]。
鋰金屬可以和其他金屬在室溫下合金化,這些合金負極材料具有遠超石墨負極(372 mAh/g)的理論比容量,但是當鋰沉積容量較高時存在枝晶生長和體積膨脹等問題,因此許多研究致力于探究鋰沉積行為的影響因素和限制脫嵌鋰過程中合金負極體積膨脹的方法。研究表明,通過合金負極材料本身改性與結構設計可以解決這些問題。比如,合金化改性(如摻雜特定的合金元素),一方面可以改善負極材料親鋰性,提高鋰金屬沉積吸附能,降低鋰金屬形核過電位[4],從而實現電荷均勻分布,促進鋰的均勻致密沉積;另一方面,元素摻雜使負極表面的鋰沉積過程轉變為固溶行為,徹底解決了鋰枝晶形成的問題。此外,通過細化晶粒、構造更多的晶體缺陷可以構建快速的鋰傳輸途徑,從而提高合金負極材料的擴散系數,從動力學上促進鋰在負極內部沉積,同時抑制鋰枝晶的形成[5]。針對體積膨脹問題,近來在合金納米化、表面包覆等傳統改性方法的基礎上,提出了合金三維導電框架[6]、合金納米顆粒自發形成核殼結構[7]和梯度合金化負極[8]等新型解決策略。
本文系統地分析了不同合金負極的脫嵌鋰機制,從熱力學與動力學角度分析了合金負極的鋰沉積行為,并介紹了合金負極的體積膨脹問題及其解決策略,為實現高能量密度合金負極鋰離子電池的研究提供參考。最后歸納總結了合金負極面臨的挑戰與解決方法,展望了合金負極未來的發展方向。
1 合金脫嵌鋰機制
不同合金相的脫嵌鋰反應機制不同,對于二元合金的脫嵌鋰反應,根據合金化后相態數目,可以將合金脫嵌鋰反應分為單相固溶反應和兩相反應(共晶、共析、包晶等);對于三元合金的脫嵌鋰反應,可以根據嵌鋰反應產物含鋰相組元數目分為二元相和三元相合金負極。
1.1 二元合金(Li-M)
1.1.1單相固溶反應
二元合金在嵌鋰過程中,鋰原子進入合金晶體中占據原有原子的晶格位點或填入間隙,稱為單相固溶反應。單相固溶反應前后晶體結構與組成合金的某一組元相同,晶體結構的一致性使得無新的界面能產生,降低了鋰沉積時的能壘,但界面能隨晶體結構不匹配程度的增大而增大。二元合金是否發生單相固溶反應主要依賴材料組分和反應溫度。PELTON總結了Li-Au合金相圖(圖1a)[9],兩者均存在單相固溶反應區,因此金具有良好親鋰性。鋰沉積與合金化以電勢差為驅動力,鋰金屬形核需要一定的過電位,形核過電位定義為電壓曲線中電壓平臺與電壓降最低值的差值。
崔屹等[4]報道了鋰沉積時形核過電位與相反應之間的聯系,發現嵌鋰形成富鋰合金相的單相固溶過程在電壓曲線上幾乎沒有形核過電位(圖1c)[4]。在鋰沉積過程中,鋰溶解度高的合金基體材料嵌鋰容量較大,嵌鋰過程中電極電勢下降較為緩慢,電壓曲線的斜率較小,這與鋰在金屬元素中溶解形成單相的行為相吻合;而對于Li溶解度低的合金基體材料,電壓曲線斜率相對較大,并且可以觀察到較小的鋰形核過電位(如Al為5 mV,Pt為8 mV[4]).
利用合金的脫嵌鋰單相固溶反應機制,可以篩選一定的合金元素。通常而言固溶度較大的合金負極材料具有較高的比容量,形核過電勢低,有利于實現鋰的均勻沉積,抑制鋰枝晶生長;但是由于合金材料自身物化特性不同(如固溶度、脫嵌鋰電位、密度等),對電池能量密度的影響需要具體分析。
1.1.2兩相反應
合金負極出現鋰沉積過電位一般可分為兩種情況。第一種,如Si等物質,其合金相圖表明常溫下脫嵌鋰過程幾乎不存在單相固溶區(圖1b)[10],說明合金負極嵌鋰過程中發生兩相反應生成兩相合金,其晶體結構與固溶體完全不同,成分和性能也不相同,并且與Li組元存在晶體結構差異,導致鋰沉積存在一定的形核過電位;此外,由于Si等物質熱力學行為不一致,使得這些材料中鋰的形核過電位不同(圖1d)[4]。第二種,如Au、Ag、Al、Mg等物質,其與鋰可以互溶并且當合金中鋰含量超過固溶體溶解度時出現兩相反應,在合金相圖中體現為兩相區,存在鋰沉積過電位。

圖1 合金相圖((a)Li-Au[9];(b)Li-Si[10]);(c,d)不同基底上電流密度為10 μA/cm2時鋰沉積電壓曲線[4]Fig.1 Alloy phase diagram ((a)Li-Au[9];(b)Li-Si[10]);(c,d)Lithium deposition voltage curve on different substrates at 10 μA/cm2[4]
硅具有很高的理論比容量(4 200 mAh/g),是最有潛力替代石墨(372 mAh/g)的負極材料,但是硅的脫嵌鋰過程伴隨著較大的體積變化(大約300%),許多研究致力于解決硅的體積膨脹問題[11-12],下面以硅的脫嵌鋰過程為例說明兩相嵌鋰反應過程。由鋰硅合金二元相圖[10](圖1b)可知硅在循環過程中的相變行為[13](圖2):
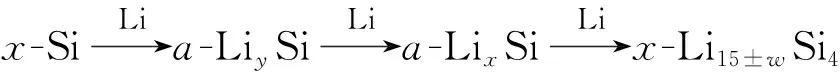
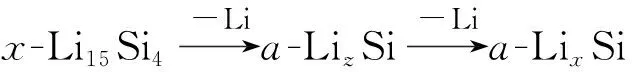
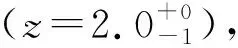
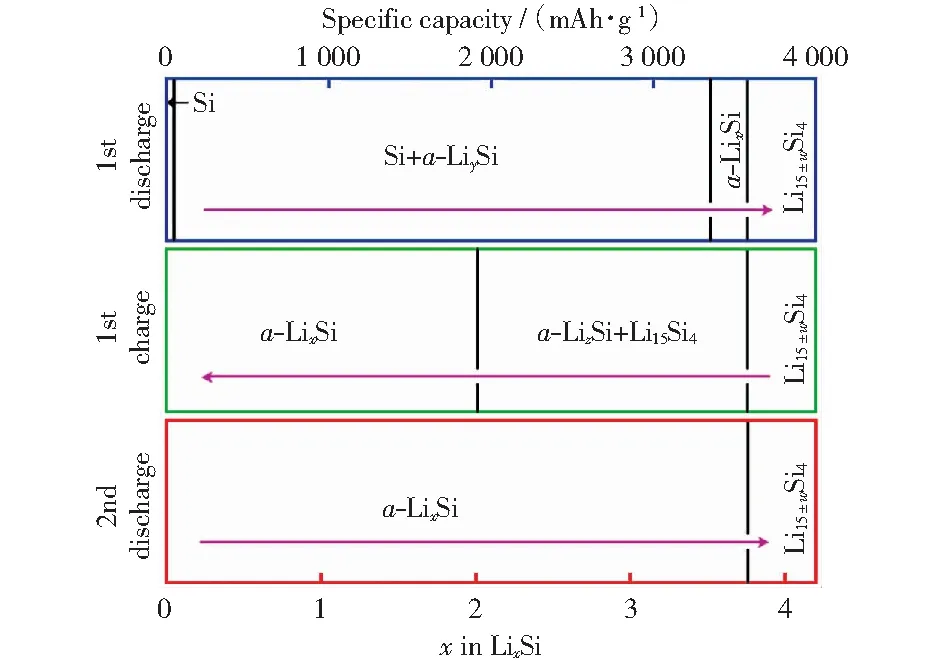
圖2 室溫下0.005~0.9 V之間Li/Si充放電循環中發生電化學反應的相轉變過程[13]Fig.2 Phase transition process of electrochemical reaction in Li/Si charge-discharge cycle between 0.005 and 0.9 V at room temperature[13]
在合金脫嵌鋰過程經歷兩相區時,反應前沿為摩爾體積相異的兩相共存區,兩相界面由于不均勻的體積膨脹導致相界面產生應力,并可能發生塑性變形和斷裂。MCDOWELL et al[14]通過理論模型描述了硅在脫嵌鋰過程中產生的界面應力及引發的塑性變形和斷裂,顆粒受損和容量衰減現象與兩相反應同時出現,因此在循環過程中需要盡量避免兩相反應[14-21]。但是也有許多研究在兩相區循環過程中獲得了較好的循環性能,如通過將合金負極材料納米化和復合化,可以避免在充放電過程中由于體積變化產生的顆粒粉化現象導致的容量衰減[11-12]。XUN et al[22]通過向晶粒大小150 nm的錫負極中添加多氟芴型導電聚合物粘結劑,使錫在粘結劑作用下兩相界面重聚合,使得錫負極在導電粘結劑作用下經過兩相區也可以展現良好的循環性能。此外,脆性合金材料同樣被發現在兩相區可以展現良好的循環性能,如BRYNGELSSON et al[23]發現Sb/Sb2O3在1 V左右有平坦的電壓平臺,而且在0~2 V充放電區間內循環50圈沒有容量衰減,較高的電壓平臺一方面避免了合金負極材料表面固態電解質界面膜(SEI)生長使活性顆粒失去接觸,另一方面減少了合金負極材料的顆粒粉化,因此使得容量衰減大幅度減小,但是較高的截止電壓會大大降低合金負極材料的比容量和電池能量密度。
1.2 三元合金負極(Li-X-M)
三元合金主要由鋰與兩類活性合金元素組成,依據活性元素不同大致可以分為Si-M系(M=H、Mg、Ca、Ag、Zn、B、C、Al、Sn等)合金和Sn-M系(M=Mg、Ag、Zn、C、Si、Sb、Bi等)合金。依據嵌鋰后金屬化合物組元數目,嵌鋰反應產物可以分為三元金屬化合物和二元金屬化合物。
1.2.1三元金屬化合物
以Si-M系合金為例,ANANI和HUGGINS[24]發現,相比于鋰硅二元合金,三元鋰硅合金具有更高的容量和更低的電極電位;KIM et al[25]采用電化學測量、X射線衍射(XRD)和俄歇電子能譜(AES)等分析方法研究了鋰離子插入Mg2Si的反應機理及電化學性質(反應式(1-3)).
Mg2Si+xLi++xe-→LixMg2Si .
(1)
LixMg2Si+xLi++xe-→Li臨界Mg2Si→
Li飽和Mg2Si+Mg+Li-Si合金 .
(2)
Mg+xLi++xe-→Li-Mg合金 .
(3)
首先,鋰插入到Mg2Si反螢石結構的八面體位置,形成LixMg2Si相(式1).當嵌鋰量達到臨界值,Li臨界Mg2Si分解為Li飽和Mg2Si、Mg和Li-Si合金(式2).最終,嵌入的鋰離子與鎂形成Li-Mg合金(式3).
類似的嵌鋰過程生成三元金屬化合物的合金負極還包括Si-Ag、Si-Zn、Si-Al、Sn-Mg、Sn-Ag等。Si-Ag合金在0.7 V時與鋰反應形成Li8Si5Ag3相,在更低的電壓下形成Li-Ag相;當Ag的原子百分比小于34%時,低壓下形成Li15Si4;當Ag的原子百分比大于57%時,循環過程中不會出現Li15Si4相[20]。Si-Zn合金(Li2SiZn)在嵌鋰過程中鋰插入準層狀結構中,保持著六方結構,但是當達到Li3SiZn時,層間距擴大,循環性能變差;雖然其體積比容量(550 Ah/L)略低于石墨,但其體積膨脹(2%)遠小于石墨[26]。Si-Al合金與鋰共同形成三元合金相(Li-Al-Si合金),嵌鋰經過兩相反應生成Li7Al3Si4;由于兩相反應機制,Li-Al-Si的循環性能很差[27]。TILLARD et al[28]研究了Li5AlSi2和Li9AlSi3的電化學性質,發現相比于Li-Al-Si合金,Li9AlSi3在LixAlSi3(4.25≤x≤11)單相區可以獲得可逆的循環容量,而Li5AlSi2的循環性能仍然比較差。
1.2.2二元金屬化合物
同樣以Si-M系合金為例,WOLFENSTINE[29]報道了Ca2Si負極脫嵌鋰機制并發現其容量與其顆粒大小有關,同時發現高模量、低延展性顆粒有助于抑制容量衰減。根據Ca-Si-Li三元相圖,Ca2Si負極容量嵌鋰后生成鋰鈣和鋰硅合金,脫嵌鋰機制如下:
10.8Li+CaSi2→Li2Ca+2Li4.4Si .
(4)
通過類似的嵌鋰過程生成二元金屬化合物的合金負極還包括Li-Si-C、Li-Si-Sn、Li-Sn-Zn、Li-Sn-Al、Li-Sn-Bi、Li-Sn-Sb等。如Si-C負極,一般硅顆粒表面利用碳包覆作為導電薄層,鋰可以分別與碳和硅發生合金反應,生成Li-C和Li-Si兩個二元金屬化合物。Si-Sn合金具有較大的質量/體積比容量,如BEAULIEU et al[30]利用磁控濺射制備的Si0.66Sn0.34非晶復合薄膜可逆容量接近1 800 mAh/g,平均脫鋰電位為0.5 V,其體積比容量約2 160 Ah/L,體積膨脹率約264%,并推測Li/a-LixSi0.66Sn0.34半電池電壓-容量曲線的充放電遲滯是由于脫嵌鋰過程中鋰原子周圍的局部原子(Si、Sn)環境發生變化所耗散的能量所致。HATCHARD et al[31]同樣采用磁控濺射制備了硅錫(Si1-xSnx,x=0~0.45)非晶復合薄膜,表現出良好的電化學性能,在循環伏安曲線中未看到尖峰,說明脫嵌鋰過程是連續且均勻的,錫的添加提高了可逆容量(3 500 mAh/g)和容量保持率(>99%每圈)。磁控濺射方法用于合金化負極的制備具有巨大潛力,但是,面對商業化生產許多實際問題尚待解決。
2 合金負極鋰離子電池研究進展
2.1 鋰離子與合金負極的鋰化反應研究
鋰離子與合金負極發生鋰化反應實現鋰離子的存儲,鋰化行為主要包括合金固溶反應和均膜狀沉積兩種類型。鋰離子與合金負極反應的熱力學特性主要體現在親鋰性、吸附能等,合金具有良好的親鋰性、較大的吸附能等,有助于降低鋰沉積能壘,促進鋰均勻沉積;如果鋰金屬與合金負極反應具有優異的動力學特性,將有助于鋰在合金體相內擴散,形成均一的相或穩定的膜狀沉積,使得合金負極具有優異的倍率和循環性能。
2.1.1合金負極熱力學特性
根據合金負極的熱力學特性,與鋰金屬晶體結構相近且與鋰具有一定溶解度的金屬元素具有良好的親鋰性,在鋰沉積過程中具有極小的形核過電位,能夠減少空間電荷分布不均勻的現象,從而促進鋰的均勻沉積。特別地,隨著金屬元素與鋰固溶度的增加,其形成的合金負極理論比容量提高。LIU et al[32]采用直接電沉積法在三種不同集流器(銅、聚吡咯、鈦)表面沉積了銅錫合金作為負極,組裝的電池具有良好的循環性能和較高的體積比容量,其中聚吡咯由于導電性較差,銅錫合金結晶度不佳,電池倍率性能并未得到提高,而以鈦板為銅錫合金電化學沉積基底的負極材料具有優異的電化學性能。
此外,利用一些金屬與鋰互溶的特性可以調節鋰沉積行為,許多研究以這類合金元素作為晶種,提供了鋰優先沉積的位點,提出了一種選擇性沉積的策略。YAN et al[4]研究了鋰與其他金屬合金化過程對鋰形核過電位的影響,發現當利用金作為鋰沉積的晶種時,即使在晶種覆蓋率較低的情況下也可以獲得較理想的鋰選擇性沉積。根據此原理制備了內部含有金納米顆粒的碳球殼,組成的對稱電池在0.5 mA/cm2的電流密度和1.0 mAh/cm2的面積容量下可以循環300圈(1 200 h);YUAN et al[33]設計了由NaMg(Mn)F3@C核殼微型結構構成的“膠囊”作為鋰金屬負極的載體,活性組分由電解液、可溶性堿金屬氟化物與不溶性過渡金屬氟化物混合組成,選擇碳作為保護殼層能夠進一步降低氟化物的溶解速率,長期釋放功能金屬離子和氟離子,在銅箔表面原位形成功能金屬層,誘導鋰的均勻沉積。溶解于電解液的氟離子,在沉積表面生成LiF,提高了負極穩定性,由此構成的合金負極能夠在2 mA/cm2和1 mAh/cm2電流密度下穩定循環1 000次,并保持高達98%的庫侖效率。
除了添加固溶合金元素可以提高親鋰性外,WANG et al[34]發現合金元素與鋰之間形成的化學鍵強弱和熔融鋰與基質反應的吉布斯自由能對鋰與基質潤濕性起決定作用。如鋰與松香酸的化學吸附能比鋰與銅簇的吸附能高,形成了更強的化學鍵,提高了潤濕性。他們還研究了有機涂層改性基底和熔融鋰改性合金元素對熔融鋰潤濕性的影響,發現含-COOH、-OH、-SO3H、-NH2等有機涂層的基底和添加Mg、In、Ca、Sr、Ba等元素,可以提高對鋰的潤濕性;同時發現表面吸附能也對鋰沉積行為存在影響,如在鋰鎂合金負極體系中,當鋰在負極表面沉積時,由于鋰鎂合金較好的親鋰性和較大的表面能,使得鋰沉積形態呈膜狀。CHOI et al[35]通過密度泛函理論(DFT)計算發現,不同晶面對鋰的吸附能不同。在不同直徑鋰納米棒中,{100}晶面邊緣吸附能最小,并且始終小于純鋰{100}晶面吸附能(-1.256 eV),因此鋰優先在已沉積鋰的{100}晶面邊緣沉積生長,形成結構疏松的枝晶鋰。而鋰鎂合金表面吸附能(-1.324 eV)小于鋰納米棒任意晶面處的吸附能,鋰優先在負極表面沉積,形成致密的膜狀沉積層(圖3a、b).
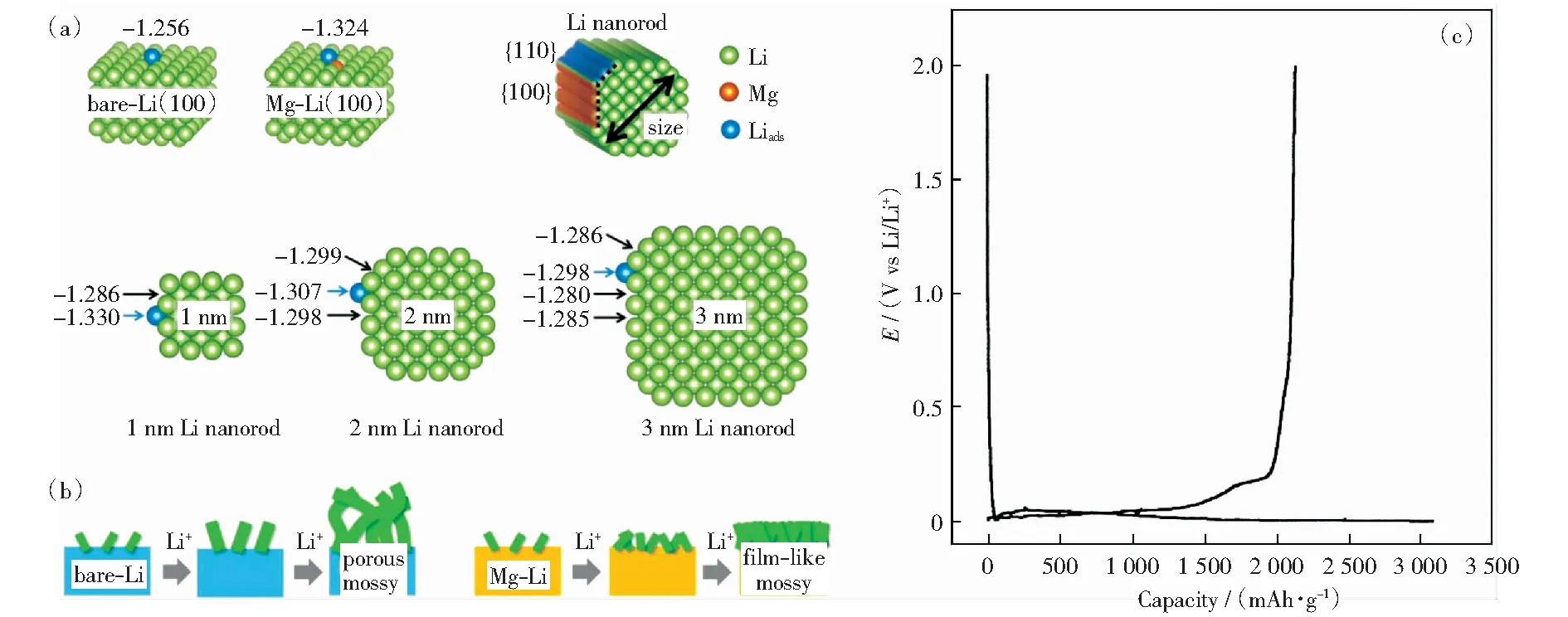
圖3 (a)鋰原子在不同金屬表面的吸附能(eV)(對于枝晶,最優先吸附標記為藍色);(b)純鋰和Mg-Li合金表面鍍鋰行為示意圖[34];(c)鎂電極在5 mA/g電流密度下的電壓曲線[35]Fig.3 (a)Adsorption energies (eV) of differnent metal surface(For the whiskers, the most energetically preferred adsorption is marked with blue spheres); (b)Schematic representations of Li plating behaviors on the bare Li and Mg-Li surfaces[34]; (c)Voltage curve of magnesium electrode at 5 mA/g current density[35]
合金負極改性對抑制鋰枝晶起到了良好的效果,許多工作通過測試大電流密度和高沉積面容量下鋰鋰對稱電池的電化學性能來研究合金負極抑制枝晶生長的能力。合金負極匹配不同的電解質體系(固態電解質、液態電解質)表現的電化學性能需要分別進行對比(圖4)[37-52]。經過對比不同鋰沉積行為,發現鋰在負極內部沉積的電池循環性能比鋰沉積形成膜狀結構時的電池性能更加優異[6,8-37]。
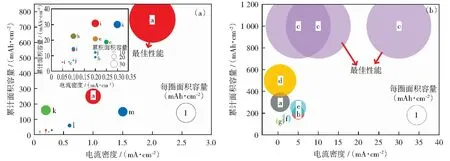
參考文獻包括:圖4a:a) [37], b) [38], c) [39], d) [40], e) [41], f) [42], g) [43], h) [44], i) [45], j) [46], k) [47], l) [48];圖4b:a) [4], b) [35], c) [6],d) [49],e) [50], f) [51], g) [52]圓面積大小代表每圈的面積容量;每項工作都被繪制成相同顏色的點圖4 (a)合金負極固態電解質電池的循環性能;(b)合金負極液態電解質電池的循環性能Fig.4 (a)The cycle performance of alloy anode solid electrolyte battery; (b)The cycle performance of alloy anode liquid electrolyte battery
促進鋰在負極內部沉積需要結合合金的熱力學及動力學特性進行綜合調控,合金負極的熱力學特性決定了鋰的優先形核位置,動力學特性決定了鋰的擴散速率,除了優化鋰優先形核位置,還需要進一步提高鋰在負極內部的擴散速率,促進鋰在合金負極內部的沉積。
2.1.2合金負極動力學特性
負極動力學性質決定電極鋰離子輸運能力和反應速率,顯著影響鋰沉積行為。動力學特性具體表現為電子電導、離子電導、合金負極擴散系數和界面阻抗等性質。堿土金屬元素通常與鋰具有一定的固溶度,例如常溫下鎂在鋰中具有較大的固溶度(68%),因此鋰鎂合金具有較高的理論比容量(3 070 mAh/g,Li2.78Mg),鎂作為合金負極的組分被廣泛研究,但是其存在動力學特性較差、空氣反應活性較強等缺點,限制了其實際應用。
首先,鋰鎂合金作為負極存在著界面阻抗高、易產生枝晶和界面穩定性低的問題,在大多數電解液體系中,鎂電極表面會自發形成一層表面膜,導致其表面阻抗高幾個數量級;同時,由于鎂電極表面形成的氧化膜和SEI膜,限制了動力學特性,導致其倍率性能較差,最大嵌鋰電流密度僅為10 mA/g[36].其次,如圖3(c)所示鎂具有極低的脫嵌鋰電位(嵌鋰:15 mV;脫鋰:50 mV),更容易使鎂成為沉積鋰的等勢體,結合其擴散系數較低的特性,使得鋰易在電極表面沉積,而非溶入電極體相內形成固溶體,導致鋰枝晶的生長。在固態電池體系中,鋰在負極表面沉積/剝離行為會使循環過程中負極與固態電解質界面接觸逐漸變差,導致電池容量迅速衰減。總體來說,鋰在負極體相內沉積減少了鋰金屬直接暴露在界面處的可能性,提高了界面穩定性,抑制了鋰枝晶的生長。
鋰金屬與固態電解質極差的界面接觸導致極大的界面阻抗,是在充放電過程中界面接觸性會進一步變差。JOW et al[53]組裝了Li/LiI(Al2O3)/PbI2電池來研究充放電過程中電極與固態電解質的界面變化,發現放電時界面處的鋰氧化為Li+運輸至正極,而負極與固態電解質界面處留下空位,當放電速率超過電極中鋰的擴散速率時,空位無法被及時填充,造成界面接觸被破壞。KRAUSKOPF et al[54]通過將鋰與LLZO固態電解質疊放并施加幾百兆帕的壓力發現鋰/固態電解質的界面阻抗幾乎為0,表明鋰和固態電解質的幾何接觸和固態電解質的離子傳導決定了平衡狀態下的界面阻抗(圖5(a)).此外,由于空位的積累導致接觸失效和界面孔隙的形成,負極中的空位擴散速率限制了負極的倍率性能。動力學模型的結果表明,只有當電流密度不超過約100 μA/cm2時,電極/電解質的界面形態才會保持穩定,而這樣的電流密度遠達不到實際應用的要求。
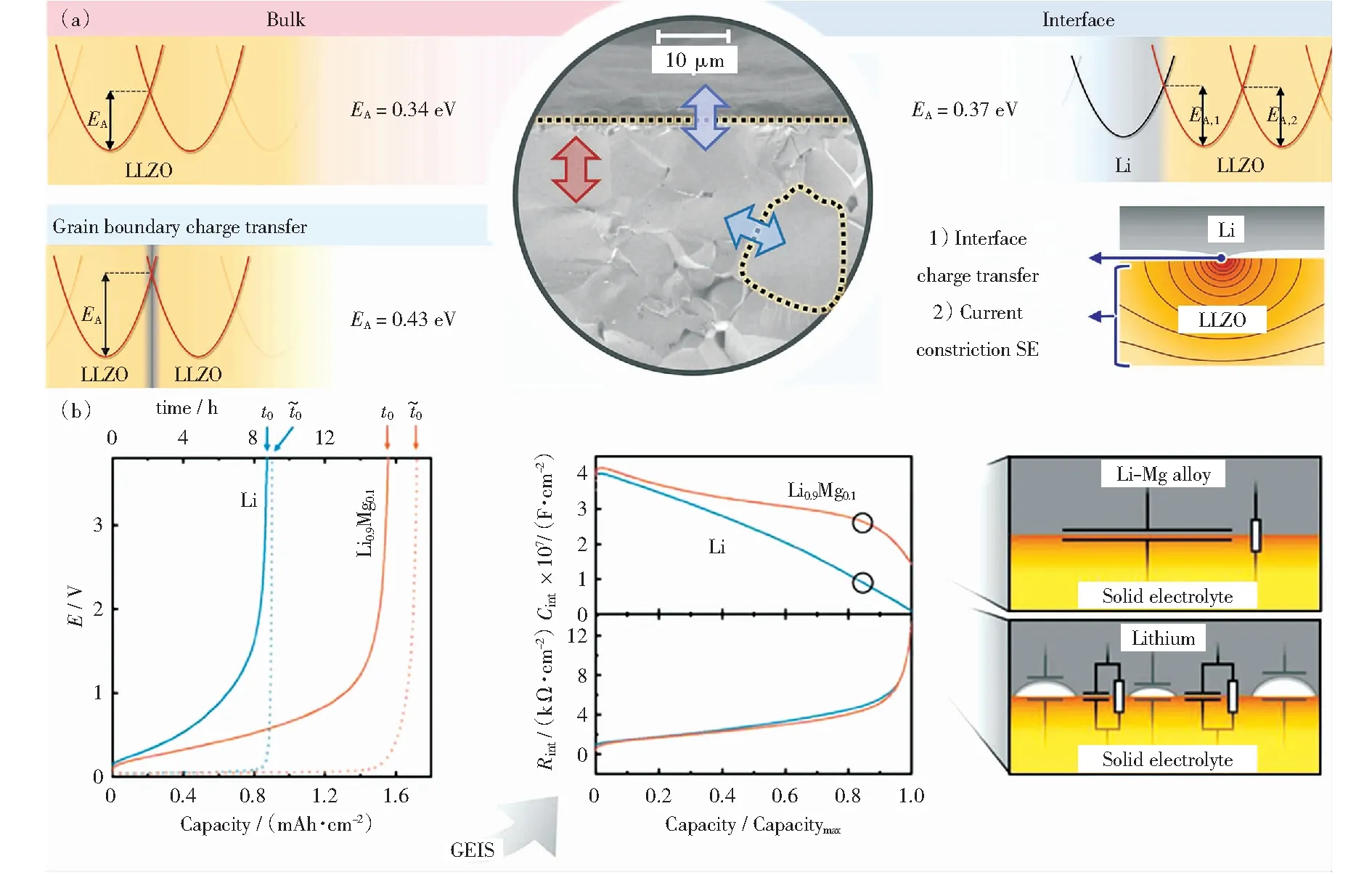
圖5 a.隨溫度變化的阻抗譜測量得到的活化能[54];b.Li和Li-Mg合金電極操作溶出實驗結果[5]Fig.5 a.The activation energies measured with temperature-dependent impedance spectroscopy[54]; b.Results of operando stripping experiments of Li and Li Mg alloy electrodes[5]

為了解決鋰鎂合金負極電極電位低導致的鋰枝晶生長問題,胡良兵等[37]通過簡單的熔融方式制備了鋰鎂合金負極,將鋰鎂合金熔融在石榴石型固態電解質上退火冷卻,發現鋰鎂合金可以起到既導電子又導離子的雙導電框架作用,組裝得到的對稱電池在1 mA/cm2和1 mAh/cm2條件下循環250圈,緊接著在2 mA/cm2和2 mAh/cm2條件下循環250圈,累計循環1 000 h后未出現明顯極化增大,并且與固態電解質界面阻抗小于50 Ω/cm2.雙導電框架結構的設計誘導鋰在合金負極體相內沉積,解決了鋰鎂合金電極電勢低、易生長枝晶的問題。此外,王成林等[52]在金屬鋰箔襯底上磁控濺射制備了74 nm厚的Al薄膜,系統研究了Li-Al合金保護層的作用,發現合金保護層具有不同于純電子導體和純離子導體保護層的特點,該混合導體保護層使鋰原子在合金層表面還原,然后借助化學勢梯度擴散進入鋰合金保護層,通過嵌鋰使合金層生成了Li9Al4等合金相,避免了金屬鋰在合金層表面的沉積,有效抑制了鋰枝晶和副反應的產生,合金混合離子/電子導電層保護機理為無枝晶合金負極的開發提供了新的解決策略。因此,發展合金負極是解決與固態電解質界面接觸較差和穩定性問題的重要途徑。
2.2 體積膨脹解決策略
使用比容量大的負極材料(比如鋰、硅)可以顯著提高鋰離子電池的能量密度,但其在循環過程中因體積變化過大而導致的機械不穩定性等問題阻礙了實際應用。目前主要的解決方法有預留體積膨脹空間(合金雙導電框架、核殼結構)、表面包覆和納米化[6]。
合金導電框架,指一些合金元素與鋰自發進行合金化反應,一定程度地脫鋰后成為既導電子又導離子的合金負極三維雙導電框架,在調控鋰沉積的同時,一定程度上抑制體積膨脹。YE et al[56]通過制備納米鋁包覆三維納米銅集流體的納米復合結構誘導金屬鋰在電極內部均勻沉積,實驗結果表明,鋰離子得電子被還原,優先與鋁發生合金化反應,形成親鋰的鋰鋁合金層,之后鋰鋁合金層作為鋰的形核位點,調控鋰沉積行為,組裝得到的對稱電池和匹配磷酸鐵鋰的全電池具有良好的電化學性能。孫永明等[6]利用自發的合金化反應,通過在常溫下將鋰箔和錫箔簡單重復壓延和折疊,制備出一種互穿型鋰金屬/鋰錫合金(Li/Li22Sn5)負極,將面容量為15 mAh/cm2的Li/Li22Sn5負極與4 mAh/cm2的三元正極進行匹配(N/P=3.75),并在4 mA/cm2的電流條件下進行測試,NCM‖Li/Li22Sn5全電池首次循環表現出3.33 mAh/cm2的容量,并在100個循環之后電池容量依然保持為2.80 mAh/cm2,容量保持率為82%,體現了合金負極巨大的商業應用潛力。KIM et al[57]進一步推進了合金化負極的商業應用,使用超薄的Ag-C層負極,鋰源全部來源于正極,其中Ag降低了鋰沉積過電位,嵌鋰后,Ag-C合金負極自發分層形成合金化Li-Ag層與含少量Ag的碳層,分隔鋰與固態電解質并阻止SEI層形成,同時保持了穩定的界面接觸。使用高負載(6.8 mAh/cm2)的高鎳層狀氧化物和硫化物電解質(Li6PS5Cl)設計的0.6 Ah的袋狀電池,能量密度高達900 Wh/L,庫侖效率高達99.8%以及1 000圈的循環壽命(60 ℃).
巧妙設計的核殼結構同樣可以抑制體積變化。PARK et al[58]以修飾原子級Zn的多孔碳骨架材料(PCF-E)作為負極,嵌鋰過程中先后發生鋰沉積與合金化反應,碳骨架有效限制了合金體積膨脹,實現了均勻可逆的鋰沉積/剝離,組裝得到的Li‖PCF-E電池350圈循環后依舊保持良好性能。伍凌等[59]利用化學性質穩定、體積膨脹小的氫化TiO2(H-TiO2)作為包覆層,負載合金型負極材料SnS2,SnS2內豐富的Ti-S鍵被固定在TiO2支撐體中,利用支撐體中氧缺陷提高了其與SnS2及其放電產物的結合力,有效地抑制了SnS2在長期充放電過程中與支撐體的剝離和團聚。
此外,納米化能夠有效解決合金負極體積變化導致的活性顆粒粉化或失去接觸的問題。CHO et al[12]提出了硅納米顆粒體積膨脹臨界尺寸的概念,當硅納米顆粒的尺寸<10 nm時,其體現出晶相與無定形相共存的性質,在脫嵌鋰過程中保持原有的顆粒大小不變。由于活性顆粒尺寸達到納米級別,表面積與體積比急劇增大,位錯會迅速地遷移至表面,因此小于20 nm的鋰硅納米顆粒或小于100 nm的納米線的斷裂韌性顯著提高[60]。由于納米顆粒巨大的表面積會形成大量的SEI,使納米負極顆粒一般具有較低的初始庫侖效率(ICE),使用碳包覆可以有效提高電池的循環壽命和庫侖效率。MAGASINSKI et al[60]設計了一種大規模的自下而上的納米尺度硅組裝策略(圖6(a)),通過化學氣相沉積(CVD)法將硅納米顆粒均勻分散在退火導電炭黑上,再將其組裝成剛性球狀顆粒,顆粒內不規則的通道提供了鋰離子快速遷移路徑,并且預留了體積膨脹空間,獲得了超高的可逆比容量(1 950 mAh/g).LIU et al[61]通過微乳化法合成了直徑500 nm~10 μm的石榴結構硅碳顆粒(圖6(b)),通過硅顆粒的納米化防止材料粉化,碳包覆提供了快速的電子輸運通道,保留硅的內部空間,抑制了體積膨脹并限制SEI僅在外表面形成。此外,由于微結構降低了電極與電解質接觸面積,獲得了較高的庫侖效率(99.87%)和體積比容量(1 270 mAh/cm3).ZHAO et al[62]設計了一種致密的石墨/硅/碳(GSC)復合材料,通過砂磨硅顆粒可以同時調節硅的粒徑和氧化程度,獲得粒徑約為125 nm、氧化層(SiOx)厚度約為8 nm的納米顆粒,并均勻地涂覆在石墨表面。納米晶粒與SiOx氧化層的協同作用可以使體積膨脹最小化,制備得到的高振實密度(0.95 g/cm3)的GSC,初始可逆容量為675.0 mAh/g,當充電倍率增加到10 C時,GSC仍能提供528.0 mAh/g的容量。因此,納米化也是一種有效解決合金類負極循環穩定性的方法,但是需要考慮其與電解液較大的副反應問題,尤其是首效較低的問題。
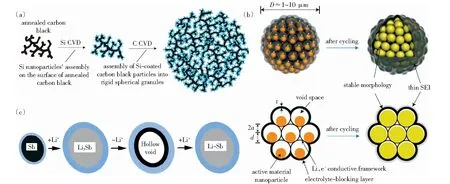
圖6 (a)自下而上的納米尺度的硅碳顆粒組裝策略[60];(b)石榴結構硅顆粒制備過程示意圖[61];(c)銻納米晶體脫嵌鋰機制[7]Fig.6 (a)Bottom-up nanoscale silicon-carbon particle assembly strategy[60]; (b)Schematic of the fabrication process for silicon pomegranates[61]; (c)De-intercalation mechanism of antimony nanocrystals[7]
結合核殼結構和納米化的優勢,具有特殊性質的納米合金顆粒在合金化過程中還可以自發地形成核殼結構,有效地解決了體積膨脹引發的一系列問題。BOEBINGER et al[7]通過原位透射電子顯微鏡(TEM),發現原始的Sb納米晶體為球形或橢圓形,并具有約2 nm厚的天然氧化物殼層,研究發現能否自發形成空心結構取決于晶體尺寸大小,小于30 nm的銻(Sb)納米晶體能夠在脫鋰過程中自發形成均勻的空隙,在循環過程中可逆地填充和產生空隙(圖6(c)).納米顆粒Sb的靜態外表面可以穩定SEI,獲得高的庫侖效率,Sb納米顆粒組裝電池的容量保持率明顯高于塊狀Sb,并且單分散的Sb納米晶體(大約15 nm)顯示出比較大的納米顆粒更高的平均庫侖效率。這種自發、可逆的空心化結構提供了抑制體積膨脹的新策略。
合金負極的體積膨脹容易使基體發生塑性變形或韌性斷裂,進而導致負極/電解質界面不穩定的問題,LI et al[8]通過不同鋁含量及退火狀態的鋰鋁合金箔證明了與鋰金屬強度的不匹配是導致基體晶粒變形破碎的根本原因。此外,在鋰鋁合金嵌鋰過程中,靠近電解質一側產生富鋰合金相,之后鋰鋁合金中的鋁元素在濃度梯度作用下向界面擴散,這一過程保證了合金負極的均一性和結構穩定。通過一定成分范圍內相穩定的金屬化合物證明了梯度合金化負極策略解決體積膨脹問題的可行性,并從金屬學和機械性能的角度為解決體積膨脹和枝晶生長問題提供了新的思路。
3 結論與展望
合金負極具有很高的理論比容量,相比傳統石墨負極具有很大的優勢。然而合金負極高容量的脫嵌鋰行為容易帶來枝晶生長和電極體積膨脹的問題,阻礙了合金負極的實際應用。合金負極存在多種脫嵌鋰機制,其中固溶反應機制兼具熱力學和動力學優勢,充分利用固溶反應機制可以抑制鋰枝晶的生長,避免兩相反應過程中的顆粒粉化及破裂。因此,鋰固溶度較大的合金負極有望成為理想的負極材料。此外,通過構造非平衡缺陷可以提供鋰傳輸通道,提高電極擴散系數,提升電極反應動力學,具有很大的發展潛力。對于合金負極嵌鋰時的體積膨脹問題,可以通過構建三維雙導電結構、核殼結構和顆粒微米/納米化等方法解決,其中顆粒納米化策略引發的副反應加劇等問題同樣值得關注。在固態電池的應用上,合金負極由于其與固態電解質之間擁有良好的界面接觸與界面浸潤性常用于固態電池中,以獲得更小的界面阻抗以及更穩定的電極界面,實現全固態電池的穩定運行。未來,發展對合金負極穩定的電解液及固態電解質種類和構建穩定的電解質/電極界面對合金負極性能的發揮至關重要。總的來說,合金負極替代傳統石墨負極將是高能量密度二次電池未來重要的發展方向。
猜你喜歡
艦船科學技術(2022年16期)2022-09-22 02:15:00
北京航空航天大學學報(2021年6期)2021-07-20 07:23:54
當代陜西(2020年13期)2020-08-24 08:22:02
制造技術與機床(2017年5期)2018-01-19 02:49:17
制造技術與機床(2017年11期)2017-12-18 06:47:29
金秋(2017年4期)2017-06-07 08:22:16
蘇州科技大學學報(自然科學版)(2017年1期)2017-03-20 15:25:18
中國材料進展(2016年10期)2016-12-26 06:50:20
濰坊學院學報(2016年2期)2016-12-01 13:00:11
新聞傳播(2015年11期)2015-07-18 11:15:04
