一測多評法測定補心氣口服液中7種成分的含量
2021-02-03 03:33:00張伯鋒陳海燕濟南市中醫醫院藥劑科濟南500濟南市第五人民醫院濟南50000廣西衛生職業技術學院南寧53003
西北藥學雜志 2021年1期
關鍵詞:檢測
張伯鋒,王 杰,陳海燕(.濟南市中醫醫院藥劑科,濟南 500;.濟南市第五人民醫院,濟南 50000;3.廣西衛生職業技術學院,南寧 53003)
補心氣口服液由人參、黃芪、石菖蒲和薤白4味中藥制成,主治氣短、心悸、乏力和頭暈等心氣虛損型胸痹心痛病癥[1]。現代研究表明,補心氣口服液對慢性穩定型心絞痛[2]、慢性心力衰竭心氣虛損證[3]、心血管神經癥[4]、慢性肺心病急性發作[5]和冠心病心絞痛[6]等疾病的臨床治療效果顯著,能有效改善心肌缺血和臨床癥狀,減少心肌損傷,改善心肌供血,降低心肌耗氧和機體炎癥反應,可提高患者的生活質量等。補心氣口服液現行質量標準[1]和文獻報道[7]僅對黃芪甲苷進行了定量檢測,未對方中其他成分進行定量研究,中藥及其制劑具有成分繁多復雜、協同作用的特點,單一成分控制模式難以確保中成藥復方制劑臨床療效的一致性,多指標成分控制模式已廣泛應用于其質量控制中。一測多評(QAMS)法利用中藥成分間存在的內在函數關系,通過測定其中一種對照品穩定易得的成分,實現多指標成分含量的同時測定,降低檢驗成本,有助于多指標成分控制模式的普及應用。本研究參考中藥質量標志物確認原則,以毛蕊異黃酮葡萄糖苷、毛蕊異黃酮、芒柄花素、人參皂苷Rg1、人參皂苷Re、β-細辛醚和α-細辛醚為研究對象,選取毛蕊異黃酮葡萄糖苷作為內參物,建立QAMS法同時測定補心氣口服液中7種成分的含量,為全面評價補心氣口服液的整體質量提供科學依據。
1 儀器與試藥
1.1儀器 Waters 2695型高效液相色譜儀(美國Waters公司);Agilent 1100型高效液相色譜儀,Agilent Extend-C18色譜柱(250 mm×4.6 mm,5 μm)、Zorbax Eclipse C18色譜柱(250 mm×4.6 mm,5 μm),美國Agilent公司;Diamonsil C18色譜柱(250 mm×4.6 mm,5 μm),北京迪科馬科技有限公司;XS105DU型電子分析天平(瑞士Mettler Toledo公司)。
1.2試藥β-細辛醚(批號112018-201802,質量分數為99.3%)、α-細辛醚(批號100298-201203,質量分數為100.0%)、人參皂苷Rg1(批號110703-201933,質量分數為93.4%)、人參皂苷Re(批號110754-202028,質量分數為93.9%)和毛蕊異黃酮葡萄糖苷對照品(批號111920-201606,質量分數為97.6%),均購于中國食品藥品檢定研究院;毛蕊異黃酮(批號PRF8062601,質量分數為99.6%)和芒柄花素對照品(批號PRF8091225,質量分數為99.9%),均購于成都普瑞法科技開發有限公司;乙腈和甲醇為色譜純,均購于天津市彪仕奇科技發展有限公司;其余試劑均為分析純;補心氣口服液(規格:10 mL·支-1;批號:190108,190509,190604),購于湖北福人金身藥業有限公司。
2 方法與結果
2.1混合對照品溶液的制備 精密稱取7種成分對照品各適量,分別用甲醇制成7種成分質量濃度分別為0.162,0.318,0.746,0.372,1.294,2.956,0.978 mg·mL-1的單一對照品溶液;精密吸取上述7種單一對照品溶液各2.5 mL,用甲醇定容至50 mL量瓶中,制成各成分質量濃度分別為8.1,15.9,37.3,18.6,64.7,147.8,48.9 μg·mL-1的混合對照品溶液。
2.2供試品溶液的制備 精密吸取補心氣口服液2.0 mL,用甲醇定容至25 mL,過濾,即得。
2.3陰性樣品溶液的制備 按照《中國藥典》2015年版一部項下補心氣口服液質量標準的工藝處方,分別制備缺人參、黃芪和石菖蒲的陰性樣品,再按照2.2項下方法制成陰性樣品溶液。
2.4色譜條件及專屬性實驗 色譜柱:Agilent Extend-C18色譜柱(250 mm×4.6 mm,5 μm)。流動相:乙腈(A)-1 mL·L-1磷酸(B),梯度洗脫(0~9.0 min,16.0%A;9.0~15.0 min,16.0%A~24.0%A;15.0~28.0 min,24.0%A~52.0%A;28.0~42.0 min,52.0%A~74.0%A;42.0~50.0 min,74.0%A~16.0%A)。流速:1.0 mL·min-1;檢測波長分別為203 nm(0~15.0 min檢測人參皂苷Rg1和人參皂苷Re)[8-12],257 nm(15.0~50.0 min檢測毛蕊異黃酮葡萄糖苷、毛蕊異黃酮、芒柄花素、β-細辛醚和α-細辛醚)[13-18];進樣量:10 μL;柱溫:25 ℃。精密吸取2.1項下制備的混合對照品溶液、2.2項下制備的供試品溶液和2.3項下制備的陰性樣品溶液各適量,按照上述色譜條件進樣檢測,色譜圖見圖1。由圖1可知,供試品色譜圖中7種成分色譜峰峰形對稱,理論塔板數按照各成分色譜峰計算均不低于3 000,主成分峰與相鄰色譜峰分離度均大于1.5,陰性樣品對補心氣口服液中7種成分的同時測定無干擾。

圖1 HPLC圖
2.5線性關系考察 分別精密吸取2.1項下制備的7種單一對照品溶液各0.1,0.5,1.0,1.5,2.0,2.5 mL,用甲醇制成6個系列質量濃度的混合對照品溶液,按照2.4項下色譜條件進樣測定7種成分的峰面積,以質量濃度為橫坐標(x)、峰面積為縱坐標(y),進行線性回歸,結果見表1。

表1 各成分線性關系
2.6精密度實驗 取2.1項下制備的混合對照品溶液,重復進樣6次,測得7種成分峰面積的RSD值分別為1.29%,1.09%,0.96%,1.11%,0.62%,0.57%,0.79%,表明儀器精密度良好。
2.7重復性實驗 取同一批補心氣口服液,按照2.2項下方法平行制備6份供試品溶液,按照2.4項下色譜條件進樣檢測7種成分的峰面積,外標法(ESM)計算得7種成分含量的RSD值分別為1.78%,0.63%,1.39%,1.55%,1.02%,0.87%,1.14%,表明實驗重復性良好。
2.8穩定性實驗 取2.2項下制備的同一份供試品溶液,于制備后0,2,4,6,12,18 h進樣檢測7種成分的峰面積,結果7種成分峰面積的RSD值分別為1.04%,1.12%,0.93%,1.09%,0.66%,0.59%,0.82%,表明供試品溶液在18 h 內穩定性良好。
2.9回收率實驗 取含量已知的補心氣口服液9份,每份精密吸取1.0 mL,置于25 mL量瓶中,按照《中國藥典》2015年版四部要求,分別按已知含有量的50%,100%,150% 3個水平精密加入混合對照品溶液(7種成分質量濃度分別為0.057,0.119,0.272,0.138,0.487,1.061,0.364 mg·mL-1)1.0,2.0,3.0 mL,各3份,再按照2.2項下方法制成加樣供試品溶液,按照2.4項下色譜條件進樣檢測7種成分的峰面積,計算得7種成分的平均回收率(RSD)分別為97.44%(1.14%),99.80%(0.75%),100.05%(0.68%),96.98%(0.90%),98.87%(1.35%),99.88%(0.73%),97.91%(1.49%)。
2.10相對校正因子(RCFs)的計算及耐用性考察
2.10.1RCFs的計算 RCFs的計算公式:
fs/i=fs÷fi=(Xs×Yi)÷(Xi×Ys)
(1)
待測成分i質量濃度計算公式:
Xi=(Xs×Yi)÷(fs/i×Ys)
(2)
式中fs/i表示待測成分i的RCFs,X表示質量濃度,Y表示峰面積,s表示內參物,i表示其他待測成分。
精密吸取2.5項下制備的6個系列質量濃度的混合對照品溶液,進樣檢測7種成分的峰面積,以毛蕊異黃酮葡萄糖苷作為內參物,按照公式(1)計算其他6種成分的RCFs,結果見表2。
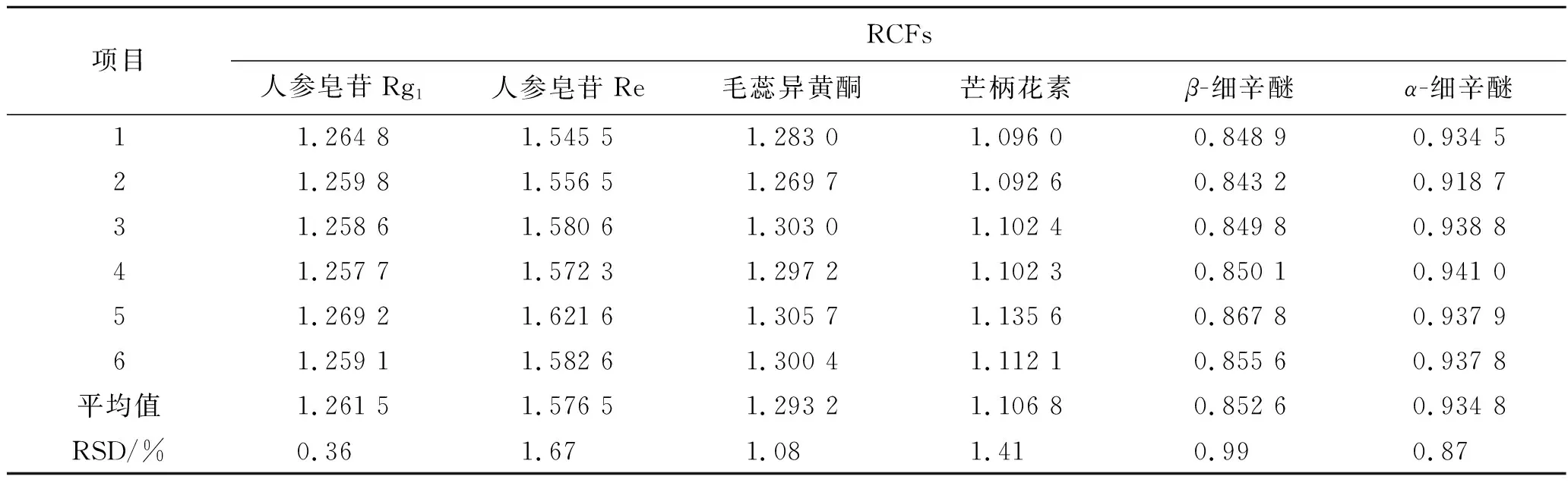
表2 各成分的RCFs
2.10.2不同儀器、色譜柱對RCFs的影響 精密吸取2.1項下制備的混合對照品溶液,按照2.4項下色譜條件進樣檢測7種成分的峰面積,考察Agilent 1100型、Waters 2695型高效液相色譜儀和Agilent Extend-C18(250 mm×4.6 mm,5 μm)、Zorbax Eclipse C18(250 mm×4.6 mm,5 μm)、Diamonsil C18(250 mm×4.6 mm,5 μm)色譜柱對RCFs的影響,見表3。
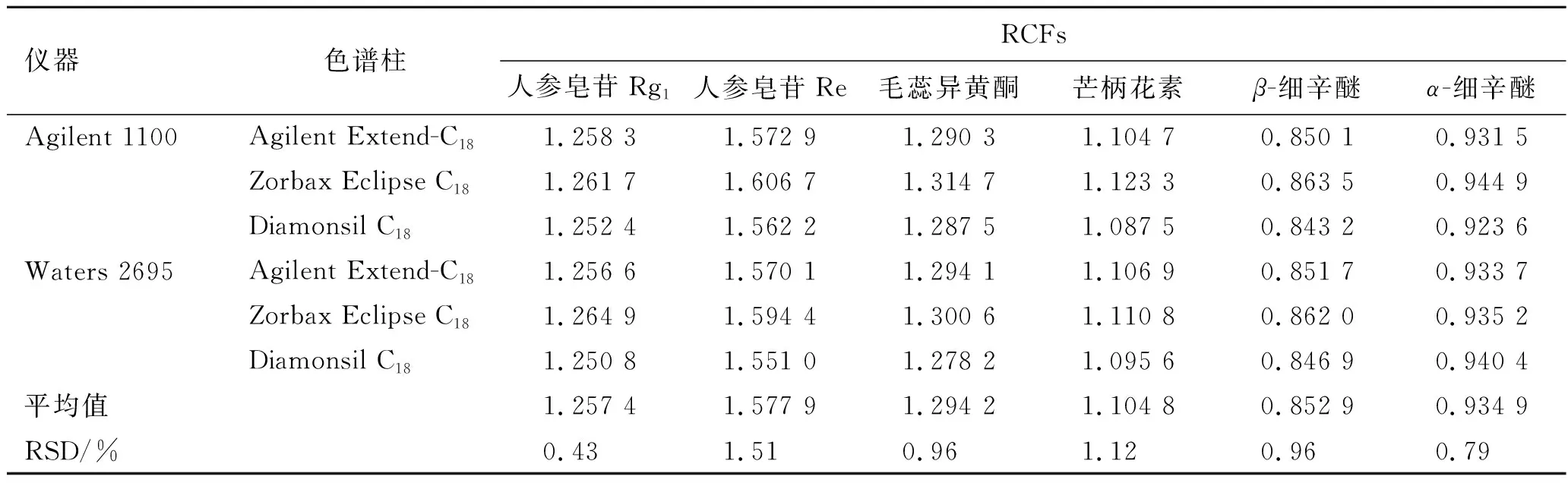
表3 不同儀器、色譜柱對RCFs的影響
2.10.3不同柱溫對RCFs的影響 精密吸取2.1項下制備的混合對照品溶液,按照2.4項下色譜條件進樣檢測7種成分的峰面積,考察不同柱溫對RCFs的影響,見表4。
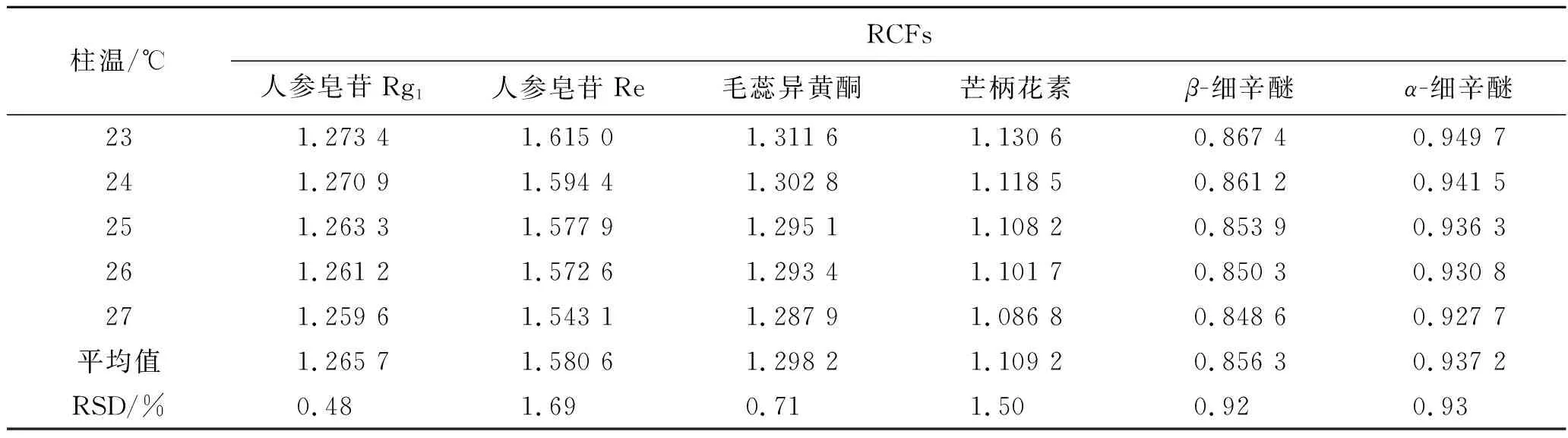
表4 不同柱溫對RCFs的影響
2.10.4不同流速對RCFs的影響 精密吸取2.1項下制備的混合對照品溶液,按照2.4項下色譜條件進樣檢測7種成分的峰面積,考察不同流速(0.8,0.9,1.0,1.1,1.2 mL·min-1)對RCFs的影響,見表5。
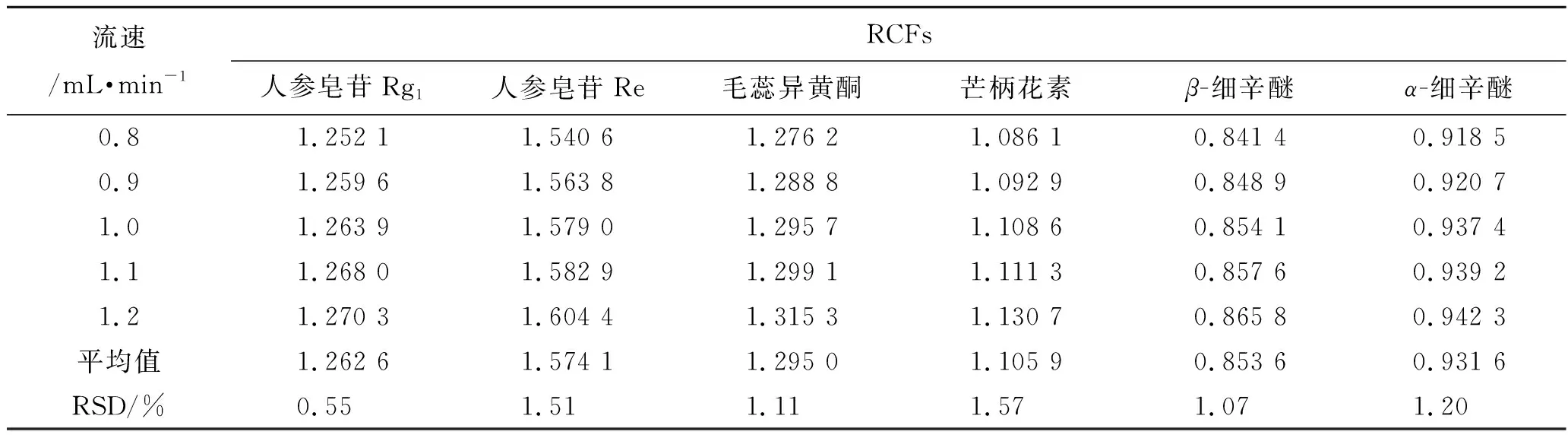
表5 不同流速對RCFs的影響
2.11待測組分色譜峰的定位 相對保留時間值法是QAMS法中待測成分色譜峰定位的常用方法。本實驗采用相對保留時間值法對待測成分色譜峰進行定位,分別記錄不同儀器和色譜柱條件下7種成分的保留時間,結果見表6。

表6 不同儀器、色譜柱對相對保留時間值的影響
2.12樣品含量測定 取3批補心氣口服液,各批次按照2.2項下方法平行制備3份供試品溶液,按照2.4項下色譜條件進樣測定7種成分的峰面積,首先采用ESM法計算7種成分的含量,再以毛蕊異黃酮葡萄糖苷作為內參物,按照2.10.1項下RCFs和公式(2)計算其他6種成分的含量,結果見表7。由表7可知,QAMS法計算值與ESM法實測值無明顯差異,相對平均偏差(RAD)均<2.00%,表明RCFs可信度較高,建立的QAMS法可同時測定補心氣口服液中7種成分的含量。
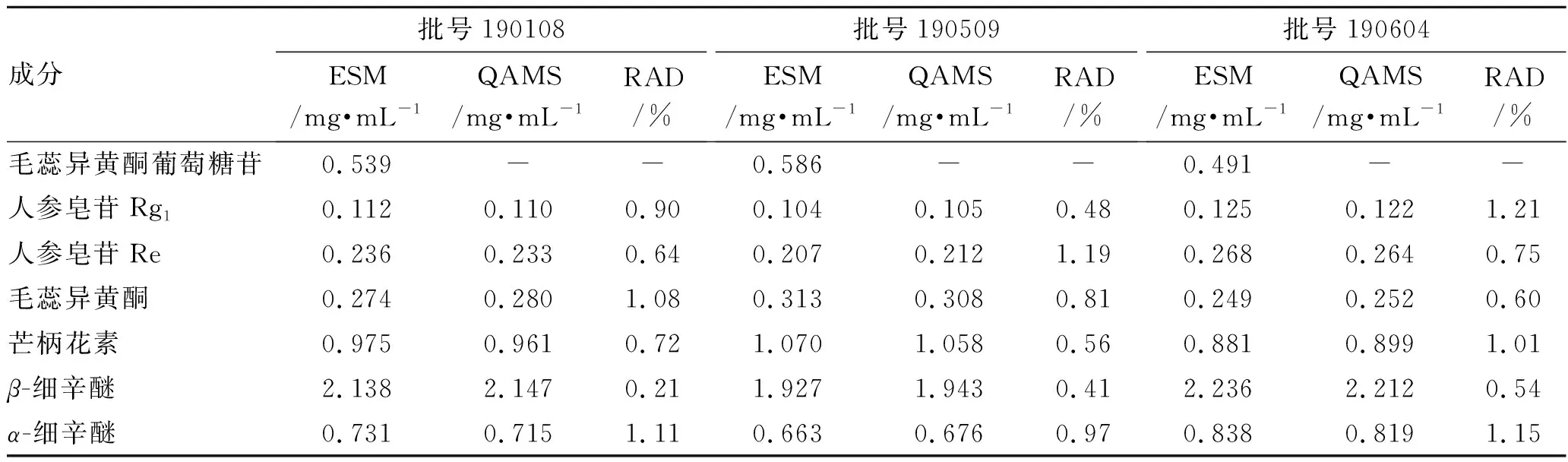
表7 7種成分的含量測定結果
3 討論
3.1目標成分的選擇 補心氣口服液由黃芪、人參、石菖蒲和薤白4味中藥材按照中醫理論配伍制成,黃芪補氣升陽、生津養血、行滯通痹,為方中君藥;人參大補元氣、生津安神,為臣藥;石菖蒲化濕開胃、開竅豁痰、醒神益智,為佐藥;薤白行氣通陽,引藥入心經,為使藥。諸藥共奏,以達補益心氣、理氣止痛的臨床功效。現代研究表明,黃芪主要含有黃酮類、皂苷類、多糖類及微量元素等活性成分,其中毛蕊異黃酮葡萄糖苷、毛蕊異黃酮、芒柄花素和芒柄花苷等為其黃酮類代表性成分[19];人參的主要藥效成分是人參皂苷,其中人參皂苷Rg1、人參皂苷Re和人參皂苷Rb1等為其代表性成分;石菖蒲主要含有β-細辛醚和α-細辛醚等揮發油類成分[20]。本文參考中藥質量標志物優選原則,選取補心氣口服液方中藥物所含毛蕊異黃酮葡萄糖苷、毛蕊異黃酮、芒柄花素、人參皂苷Rg1、人參皂苷Re、β-細辛醚和α-細辛醚為定量測定目標成分。
3.2檢測波長的確定 取2.1項下制備的7種成分的單一對照品溶液,分別在190~400 nm波長范圍內進行紫外掃描,結果發現,毛蕊異黃酮葡萄糖苷、毛蕊異黃酮和芒柄花素在260 nm波長附近有較大吸收,且雜質干擾較少,靈敏度較高;人參皂苷Rg1和人參皂苷Re在203 nm波長處有最大吸收;β-細辛醚和α-細辛醚在257 nm波長處有最大吸收。綜合考慮7種待測成分的檢測響應值和雜質干擾程度,最終確定在203 nm波長處檢測人參皂苷Rg1和人參皂苷Re,在257 nm波長處檢測毛蕊異黃酮葡萄糖苷、毛蕊異黃酮、芒柄花素、β-細辛醚和α-細辛醚。
3.3流動相的選擇 本實驗參考相關文獻,在選擇流動相時,依次對甲醇-水[14,21]、乙腈-水[8-9]、乙腈-甲醇(9∶1)[13]3個流動相系統進行考察對比,結果發現,以甲醇-水和乙腈-甲醇(9∶1)為流動相體系檢測時,基線漂移嚴重,部分待測成分色譜峰分離不完全,可能與甲醇在低波長處有紫外吸收有關;乙腈-水流動相體系檢測色譜圖基線平穩,但β-細辛醚色譜峰存在拖尾現象,考慮加入酸類改善,待測成分人參皂苷Rg1和人參皂苷Re為紫外末端吸收,甲酸[14,21-22]和冰醋酸為有機酸,存在紫外末端吸收,影響色譜圖基線和檢測結果,而磷酸為無機酸,無紫外末端吸收,遂采用乙腈-1 mL·L-1磷酸[9]為流動相進行實驗,結果7種成分色譜峰峰形對稱,與相鄰色譜峰的分離度均大于1.5,色譜圖基線較為平穩。同時對流動相比例不斷優化,最終按照2.4項下的流動相洗脫程序對補心氣口服液中7種成分的含量進行同時測定。
4 結論
本實驗首次采用QAMS法對補心氣口服液中人參皂苷Rg1、人參皂苷Re、毛蕊異黃酮葡萄糖苷、毛蕊異黃酮、芒柄花素、β-細辛醚和α-細辛醚的含量進行同時測定,所建立的方法操作簡單、結果準確,對補心氣口服液的質量標準提升和全面評價提供了參考。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
